・有機反応機構(求核置換反応と脱離反応)
【目次】
(1) 有機反応機構の規則
有機化学は、よく「暗記の化学」であるといわれます。高校で習う有機化学では、たくさんの有機化合物の名称を暗記して、さらに様々な反応機構まで暗記しなければならないからです。有機化学は、数学や物理学が苦手でも、それほど問題なく学習を進めることができるため、高校レベルでは、暗記量が物を言う学問なのです。しかし、ただの暗記だけで十分なのは、せいぜい高校レベルまでです。大学レベルでは、「有機化学は暗記の学問である」などと言う人は、恐らく一人もいません。有機化合物の種類は5000万種を超え、さらに、それに関係する有機反応まで含めると、とても暗記だけでは済まなくなるからです。それ故に、一流の有機化学者たちは、様々な有機化合物や複雑な有機反応を、系統立てて理解するようにしています。すなわち、多くの有機化合物には官能基があるため、官能基ごとに化合物を分類すれば、有機化学の学習は非常に効率的になります。また、有機反応についても、有機化学の反応の多くは、基本的な数種類に分類できるため、学習が非常に単純化されます。有機化学の基本的な規則さえ知っておけば、新しい有機化合物や有機反応を見ても、その化合物の性質や反応の仕組みがすぐに推測できるようになるのです。
有機反応を勉強する上で、最も大事なことは、電子の動きを理解することです。ただし、ここでいう電子は、「価電子」のことです。価電子は、化学結合や物性に深く関係している電子です。有機化学は「電子の化学」であり、有機反応とは、「電子の動きによる物質の化学変化」の他ならないのです。有機化学では、価電子の動きを曲がった矢印で示します。この矢印を「両刃型矢印」といいます。両刃型矢印の使い方を学ぶことは、有機化学において、非常に重要なことです。次の図.1は、炭酸イオンCO32- の3種類の構造式と電子の動きを示したものです。
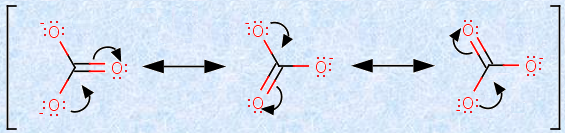
図.1 炭酸イオンCO32- の3種類の等価な共鳴構造式
有機化学では、電子の存在位置の変化を見失わないようにするために、両刃型矢印を使います。この両刃型矢印は、電子の出発地点から始まり、終着地点で終わります。両刃型矢印は、何の物理的意味も持ちませんが、式を正しく書くための工夫としては、極めて重要なものです。図.1の炭酸イオンCO32- では、酸素原子の非共有電子対と二重結合のπ電子が移動して、3種類の異なる書き方の構造式に変化します。ここで、あえて「異なる書き方の構造式」としたのは、図.1の3種類の炭酸イオンCO32- は、ただ書き方が違うだけで、すべての構造式が等価だからです。
また、図.1の構造式のどれを取っても、真の炭酸イオンCO32- を表すものではないということに注意しなければなりません。どの構造にも、炭素と水素の間には、2種類の異なる形式の結合があります。しかし、現実には、炭素と水素の結合距離は、3本ともすべて同一(0.131 nm)であることが実験的に分かっています。この距離は、正常なC=O(0.120 nm)とC-O(0.141 nm)の結合距離の中間の値です。これが意味することは、真の炭酸イオンCO32- の構造式が、ちょうど3種類の構造式の平均であるということです。真の炭酸イオンCO32- では、2個の形式負電荷は、3個の酸素原子上に均等に分散しており、各酸素原子は、2/3の負電荷を帯びることになります。このようないくつかの構造式が集まって真の構造式を作るものを、一般的には「共鳴混成体(resonance hybrid)」といいます。炭酸イオンCO32- は、3種類の「共鳴構造式(resonance form)」の間で、物理的に相互変化をするのではなく、実際には1種類の構造であり、3種類の共鳴構造式の混成体となっているのです。
また、有機化学では、曲がった片矢印も使われます。このような矢印を「片刃型矢印」といいます。片刃型矢印は、1電子の動きを示すときに使われます。両刃型矢印が2電子の動きを示すときに使われるのに対して、片刃型矢印は1電子の動きを示すときに使われるので、使い分けに十分注意する必要があります。図.2の2本の片刃型矢印は、エタンC2H6の炭素間結合が切れて、2個のメチル遊離基を生成するときの各電子の動きを示しています。

図.2 エタンC2H6の炭素間結合の開裂
(2) 求核置換反応(SN2反応とSN1反応)
典型的な有機反応には、「付加反応」・「置換反応」・「脱離反応」・「酸化還元反応」などがあります。その中でも、「置換反応(substitution reaction)」は一般的な有機反応であり、有機合成において、広く応用が可能な有機反応です。ここで例として、臭化エチルC2H5Brと水酸化物イオンOH- の置換反応を示します。
HO- + CH3CH2-Br → CH3CH2-OH + Br-
臭化エチルC2H5Brは、水酸化物イオンOH- と反応すると、臭化エチルC2H5Brのブロモ基(-Br)がヒドロキシ基(-OH)に置換され、エタノールC2H5OHと臭化物イオンBr- を与えます。このように、1つの官能基が別の官能基で置き換わる形式の反応を、置換反応と呼ぶのです。この反応において、水酸化物イオンOH- のように新たに置換してくる基を「求核剤(nucleophile)」、臭化物イオンBr- のように置換される基を「脱離基(leaving group)」と呼びます。有機化学では、それぞれの頭文字を取って、求核剤は「Nu」、脱離基は「L」と略されることが多いです。
置換反応では、共有結合の1つが切断され、それに代わる新しい共有結合が1つ形成されます。先の臭化エチルC2H5Brと水酸化物イオンOH- の反応例では、C-Br結合が切断され、その代わりに新しくC-OH結合が形成されています。この反応をもう少し詳しく見てみると、脱離するブロモ基(-Br)は、C-Br結合のσ電子2個を持ったまま脱離して、置換する水酸化物イオンOH- は、自身の非共有電子対を炭素に与えて新たにσ結合を作っているのです。ここで注意しなければならないことは、臭化エチルC2H5BrのC-Br結合を作っているσ電子は、置換反応のときにどういう挙動をするかということです。この反応では、置換するときに、σ電子ごと置き換わることに注意しなければなりません。
○(正しい) HO- + CH3CH2-Br → CH3CH2-OH + Br-
×(間違い) HO- + CH3CH2-Br → CH3CH2-OH + Br-
これを求核置換反応の一般式とすると、次のようになります。求核剤と反応基質が、ともに電気的に中性である場合は、生成物は陽電荷を帯びることになります。求核剤が陰イオンで反応基質が中性の場合は、生成物は中性になります。いずれの場合にしても、求核剤は非共有電子対を炭素に提供して、1つの共有結合を形成します。
求核剤が中性の場合:Nu + R-L → R-Nu+ + L-
求核剤が陰イオンの場合:Nu- + R-L → R-Nu + L-
求核剤は、非共有電子対を炭素に提供して、共有結合を作る物質です。それ故に、非共有電子対を持っているものならば、理論的には何でも求核剤になります。すなわち、求核置換反応においては、脱離基もまた、共有結合を形成できる非共有電子対を持つので、求核剤として働きます。つまり、求核置換反応は、原則として、すべて平衡反応になります。平衡反応については、反応速度と化学平衡でも説明していますが、競争する平衡反応は、少なくとも原理的にはすべて可逆反応であることを理解する必要があります。
しかしながら、実際的には、多くの求核置換反応は、可逆反応ではありません。その理由は、単に一方向の反応が、圧倒的に優先しているからです。なぜそうなるかについては、多くの説明が可能です。例えば、平衡にある片方の分子が、他方よりもはるかに安定である場合が考えられます。脱離基よりも強力な求核力を持った求核剤を反応で使用すれば、生成物がエネルギー的に安定になり、正反応が大きな発熱反応になって、置換反応を右辺に進行させることができます。また、生成物の1つが固体で、反応液から結晶として取り除かれたり、気体として抜けていったりする場合には、平衡定数から考えて不利な反応でも、完全に反応を一方向に進めることができます。このような条件下では、弱い求核剤を使っていて、正反応が吸熱的でも、置換反応を完結させることができます。
多くの求核置換反応の典型的な反応機構は、「SN2反応」ならびに「SN1反応」という2つの型に分類できます。ここで「SN」という記号は、「substitution(置換)」と「nucleophilic(求核的)」の頭文字を取ったものです。「2」や「1」の数字が意味する内容については、次で説明します。
(i) SN2反応
SN2反応は、次の図.3で示されるように、反応物の2分子が1段階で反応する置換反応のことです。求核剤として導入される原子や分子は、脱離していく原子の後ろ側から求核攻撃をしてきます。この反応過程の中間段階(遷移状態)では、求核剤と脱離基は、置換が起こる炭素原子を中央に挟んで結合しています。このとき、求核剤の非共有電子対の被占軌道が基質の空軌道と重なり、基質の空軌道に求核剤の電子が流れ込むに従って、脱離基がσ電子を取り込んで、炭素と脱離基の結合はだんだん弱くなっていきます。そして、求核剤と炭素との間に新しく結合が形成され始めると、炭素と脱離基の結合距離は伸びてきます。そして、炭素に結合していた他の置換基は、傘を開くように後方に曲がり、最後には立体反転して、生成物を与えるのです。
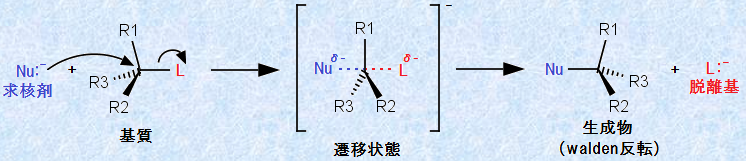
図.3 SN2反応の反応機構
「SN2反応」の数字の「2」は、この反応が2分子反応であることを示すために付けています。すなわち、SN2反応は、求核剤と基質の2つの分子が、反応機構の主要な段階に関与している2次反応なのです。SN2反応では、求核剤と基質の両方が、反応の遷移状態に関与しています。そのため、SN2反応の反応速度は、それぞれの濃度に比例します。例えば、臭化エチルC2H5Brと水酸化物イオンOH-の求核置換反応はSN2反応であるから、塩基(OH-)濃度を2倍にすると反応速度は2倍になり、臭化エチルC2H5Brの濃度を2倍にしても同じく2倍になります。ここで、反応速度定数kは、反応に固有の値であるから、SN2反応の反応速度vは、次のように表せます。
Nu- + R-L → R-Nu + L-
反応速度v=k[R-L][Nu-]
また、SN2反応では、求核剤がC-L結合の背面から攻撃してくるので、完全な立体反転を伴って反応が起こります。このことは、忘れてはならないSN2反応の重要な特徴の1つです。ラトビアの化学者であるポール・ワルデンは、SN2反応の反応機構の初期の研究において重要な貢献をしており、彼の名にちなんで、SN2反応で普遍的に見られる立体反転は、特に「ワルデン反転(Walden inversion)」と呼ばれています。
反応基質R-Lが、SN2機構で反応するとき、R基の構造は、SN2反応の速度に多大な影響を及ぼします。SN2反応は、Rがメチル基や第一級アルキル基、または第二級アルキル基の場合に限られるのです。Rがメチル基または第一級アルキル基の場合には、反応速度は速く、Rが第三級アルキル基の場合には、反応速度はゼロか、少なくとも無視できるほど小さくなります。Rが第二級アルキル基の場合には、その中間の反応速度を示します。次の表.1に、種々のR基のSN2反応におけるおおよその相対反応速度を示します。
表.1 SN2反応における平均的な反応速度比
|
R |
相対反応速度 |
|
CH2=CHCH2- |
1.3 |
|
CH3- |
1 |
|
CH3CH2- |
0.033 |
|
CH3CH2CH2- |
0.013 |
|
(CH3)2CH- |
8.3×10-4 |
|
(CH3)3CCH2- |
2×10-7 |
|
(CH3)3C- |
〜0 |
このような反応速度の順序が生じる理由は、SN2反応をよく検討すれば分ります。つまり、脱離基が結合した炭素上にアルキル基が数多く置換していると、置換反応を受ける炭素原子の背面側の混み合いが酷くなり、これが反応速度を低下させる原因になるのです。したがって、求核剤の接近に対する立体障害が特に大きな第三級アルキル化合物では、実質SN2反応は起こりません。
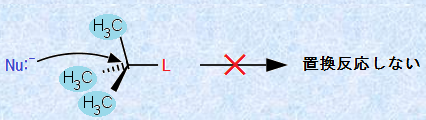
図.4 第三級アルキル化合物の立体障害
(ii) SN1反応
SN1反応は、次の図.5で示されるように、反応物の2分子が2段階で反応する置換反応のことです。初めの段階は遅く、まず反応基質がイオン化して、炭素原子と脱離基の間の結合が切れます。このときに、C-L結合を作っていたσ電子が脱離基と一緒に外れていくので、炭素陽イオンが生じます。続いて、炭素陽イオンが求核剤に捕捉される第2段階目の速い反応が起こり、炭素陽イオンは求核剤と結合して、生成物を与えます。
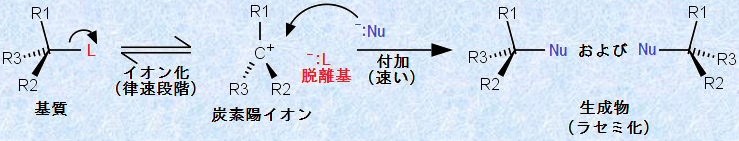
図.5 SN1反応の反応機構
「SN1反応」の数字の「1」は、2段階の反応のうち、反応速度の遅い反応段階に、反応基質だけが関与しているということを示しています。つまり、この段階に求核剤は全く関与しておらず、反応は1分子的に起こるのです。したがって、SN1反応は、以下の反応速度式で表わされる1次反応であり、反応速度は、加えた求核剤の濃度に依存しません。2段階反応の第1段階目だけが、反応速度を決定する「律速段階(rate-controlling step)」であり、この第1段階目で生じた炭素陽イオンは、速やかに求核剤と反応してしまいます。第2段階目の反応は、どのような生成物が得られるのかを決定するものの、反応速度を決定するものではないのです。
Nu- + R-L → R-Nu + L-
反応速度v=k[R-L]
また、SN1反応で、脱離基の結合している炭素が不斉な場合は、SN1反応が起こるとその結果、生成物の「ラセミ化(racemization)」が起こって、光学活性が失われることが多いです。炭素陽イオンの中心には3つの置換基しか結合しておらず、炭素陽イオンはsp2混成した電子状態を有しているので、3つの結合は平面を形成します。求核剤の攻撃は、炭素陽イオン平面のどちら側からも等しい確率で起こるので、等量のR 体とS 体のラセミ混合物が生成してきます。例をあげると、(R )-3-ブロモ-3-メチルヘキサンと水H2Oとの反応では、ラセミ混合物のアルコールが生成します。
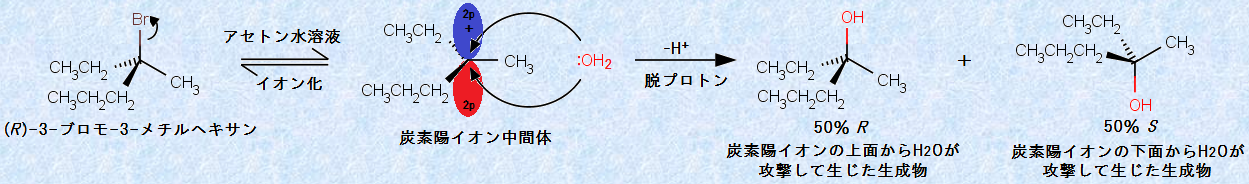
図.6 (R )-3-ブロモ-3-メチルヘキサンと水H2OのSN1反応
しかしながら、実際にSN1反応でラセミ化が起こる場合もありますが、通常はそれほど完全ではなく、立体反転した生成物が過剰に得られます。この理由は、基質から脱離基が完全に離れる前に、求核剤との反応が起こる場合が多いからです。脱離基が完全に解離しない限り、脱離基は脱離する側を遮蔽し、求核剤がこの側から攻撃して、立体保持した生成物を与えるのを邪魔します。その結果、立体反転した生成物を過剰に与えることになるのです。
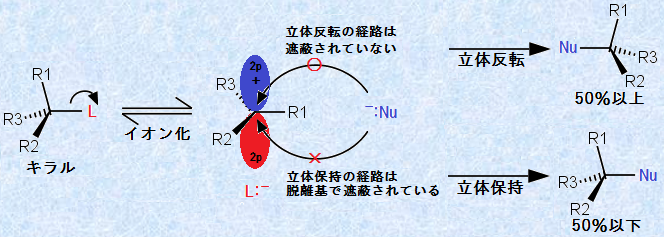
図.7 SN1反応における脱離基の遮蔽効果
また、反応基質R-Lが、SN1機構で反応するときの反応速度は、第二級アルキル化合物に比べて、第三級アルキル化合物の方がはるかに大きく、単純な第一級アルキル化合物やメチル化合物では、SN1反応を全く起こしません。SN1反応は、Rが第三級アルキル基、または第二級アルキル基の場合に限られるのです。この理由は、SN1反応の速度が、炭素陽イオン中間体の生成段階に依存していることを考えると理解できます。様々な測定結果から、炭素陽イオンの安定性は、置換基が増えるに従って大きくなることが分っています。
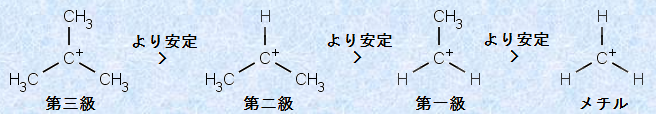
図.8 炭素陽イオンの安定性
炭素陽イオンは、空の2p軌道を持っており、隣接したアルキル基のC-H結合に使われているσ電子は、炭素陽イオンの空の2p軌道と重なることができるのです。この被占軌道と空軌道の相互作用は、炭素陽イオンの安定化をもたらします。また、この安定化は、置換基の数が増えるほど大きくなります。例として、次の図.9に、エチルカチオンCH3CH2+ の空軌道-被占軌道相互作用を示します。エチルカチオンCH3CH2+ では、1つのC-H結合が空の2p軌道と重なり、σ電子の非局在化によって、安定化を受けます。
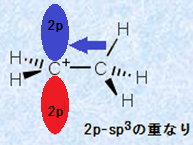
図.9 エチルカチオンCH3CH2+ の空軌道-被占軌道相互作用
すなわち、SN1反応では、炭素陽イオンの生成段階が反応速度を決定するので、反応速度の順序は、炭素陽イオンの安定性の順序に等しく、第三級>第二級>第一級となるのです。つまり、炭素陽イオンを発生させやすいものほど反応は速くなり、最も安定な第三級炭素陽イオンを生じる第三級アルキル化合物は、一般的にSN1機構で反応します。一方で、極めて不安定な炭素陽イオンを生じなければならないメチル化合物や第一級アルキル化合物は、通常SN1反応を起こしません。また、SN1反応において、第二級アルキル化合物は、第一級アルキル化合物よりも速く反応しますが、第三級アルキル化合物に比べると遅くなります。
(iii) SN2とSN1反応機構の比較
ある求核置換反応が、SN2かSN1のいずれの反応機構で進行するのかを知るためには、どうすれば良いでしょうか?先にも述べたように、反応基質R-Lにおける求核置換反応は、R基の構造に影響を受けて、どちらの反応機構で進行するのかが決まります。第一級アルキル化合物は、ほとんどSN2機構で反応するのに対して、第三級アルキル化合物は、SN1機構で反応します。第二級アルキル化合物の場合は、両方の機構で反応する可能性があります。求核置換反応がどちらの反応機構で進行するのかは、R基の構造の他には、求核剤や脱離基、反応溶媒の種類に影響を受けます。
(iii-1) 求核剤の影響
求核剤は、反応性の高い非共有電子対を持つ化学種のことで、炭素原子に非共有電子対を与えて、新しく共有結合を作る性質があります。求核剤と似たような定義として、「ルイス塩基(Lewis base)」があります(酸と塩基(酸と塩基の強さ)を参照)。ルイス塩基は、ルイス酸に非共有電子対を与えて、新しく結合を作る化学種のことです。ルイス塩基は、求核剤よりも広い定義になります。つまり、ルイス塩基の中でも、特に炭素原子と反応するものを、求核剤と呼んでいるのです。
一般的に求核性の強い求核剤ほど、炭素原子を攻撃しやすくなるので、求核性の強い求核剤は、求核置換反応においては有効になります。それでは、求核性とは、一体何によって決まるのでしょうか?優れた求核剤とは、一体どんな物質なのでしょうか?
一般的には、「塩基性が強い物質ほど、求核性が強くなる」ということができます。しかし、ここで注意しなければならないことは、塩基性が強くても、求核性の弱い物質があったり、逆に塩基性が弱くても、求核性の強い物質があったりするということです。この違いは、攻撃する炭素の空軌道に対して、求核剤が有効に作用するかどうかを考えることで理解できます。
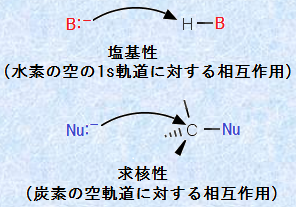
図.10 塩基性と求核性の違い
一般的に「塩基性」といった場合、それは「ブレンステッド塩基(Bronsted base)」の強さのことを意味します。つまり、「塩基性が強い物質ほど、求核性が強くなる」という表現における「塩基」とは、「ルイス塩基」ではなく「ブレンステッド塩基」のことを指しているのです。
ブレンステッド塩基性は、「水素の空の1s軌道」との相互作用の尺度であり、求核性は、「炭素の空軌道」との相互作用の尺度です。被占軌道と空軌道の相互作用は、エネルギーの近い軌道同士が重なる場合に、最も大きな安定化が得られることに注意して下さい。すなわち、求核剤の軌道が、炭素の空軌道のエネルギーに匹敵することが、求核性を決定する上で極めて重要なのです。次の表.2に、水溶液中での求核剤の相対的な求核性を示します。この中には、「強い塩基は優れた求核剤である」という法則から、大きく外れた例がいくつか見られます。
表.2 水溶液中での求核剤の相対的な求核性
|
求核剤 |
名称 |
相対的求核性 |
|
CN⁻ |
シアン化物イオン |
126,000 |
|
SH⁻ |
チオラートイオン |
126,000 |
|
I⁻ |
ヨウ化物イオン |
80,000 |
|
OH⁻ |
水酸化物イオン |
16,000 |
|
Br⁻ |
臭化物イオン |
10,000 |
|
NH3 |
アンモニア |
8,000 |
|
Cl⁻ |
塩化物イオン |
1,000 |
|
F⁻ |
フッ化物イオン |
80 |
|
CH3OH |
メタノール |
1 |
|
H2O |
水 |
1 |
例えば、シアン化物イオンCN- は、水酸化物イオンOH- よりも弱い塩基です。しかし、求核性は、約8倍もシアン化物イオンCN- の方が強いです。これは、シアン化物イオンCN- の軌道のエネルギーが、求核攻撃される炭素の空軌道のエネルギーに近いからです。
この他にも、別な要因で求核性の強さが決まることがあります。例えば、ハロゲン化物イオンの共役酸であるHX(X=F, Cl, Br, I)のpKa値から予想されるように、塩基性の強さの順はF->Cl->Br->I- です。ところが、表.2によると、求核性の強さの順は、全くこの逆なのです。ヨウ化物イオンI- は最も弱い塩基ですが、最も強い求核剤になります。また、フッ化物イオンF- は最も強い塩基ですが、最も弱い求核剤です。
このようになる理由は、水H2Oやアルコールのような極性の高いプロトン性溶媒中では、ハロゲン化物イオンは、水素結合などで強く溶媒和され、著しくかさ高くなっているからです。求核剤はかさ高くなると、反応基質に接近するのが困難になり、求核性が弱くなってしまうのです。他のハロゲン化物イオンに比べて塩基性の弱いヨウ化物イオンI- は、水H2Oやアルコールのようなプロトン性溶媒とそれほど強く相互作用をしないので、溶媒に邪魔されることなく、求核攻撃ができるという訳です。それ故に、非プロトン性溶媒であるアセトン中では、溶媒分子との水素結合による相互作用が弱くなるので、求核性の強さの順は、表.2の逆(F->Cl->Br->I-)になってしまいます。また、気相中のように溶媒がない状態でも、求核性の強さの順は逆(F->Cl->Br->I-)になります。

図.11 求核剤のかさ高さによる影響
求核性の強さは、SN2反応の速度に大きな影響を与えます。これは、SN2反応の速度が、求核剤と基質の両方の濃度に比例することを思い出せば、理解できるでしょう。つまり、強力な求核剤を用いると、SN2機構が優勢になるのです。それに対して、SN1反応では、求核剤は反応速度に影響せず、反応速度は基質の濃度のみに比例します。これは、SN1反応の速度を決める律速段階では、基質のみが関与していて、求核剤は全く反応に関与していないからです。
(iii-2) 脱離基の影響
多くの脱離基は、陰イオンとして脱離します。したがって、脱離基の陰イオンが安定になるほど、脱離基は置換されやすくなります。求核置換反応では、いずれの反応機構にしても、脱離基の陰イオンが安定であるほど、反応速度は速くなるのです。
Nu- + R-L → R-Nu + L-
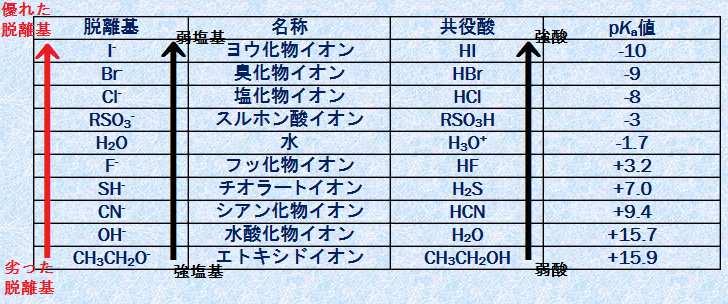
脱離基の陰イオンは、その陰イオンが弱い塩基であるほど、安定になります。つまり、その共役酸HLのpKa値と相関があるのです。pKa値は小さいほど強酸となり、その共役塩基は、弱塩基で優れた脱離基となります。別の言い方をすると、一般的に塩基性が強いほど求核性は強くなるので、「脱離基は弱い求核剤であるほど優れている」ということができます。次の表.3に、種々の脱離基をそれらの共役酸のpKa値と共に示します。ハロゲン化物イオンは、一般的に優れた脱離基であり、有機合成化学において、簡単な構造を持つ有機ハロゲン化合物(塩化物と臭化物が多い)は、多くの用途を持った原料化合物として利用されています。
表.3 種々の脱離基とそれらの共役酸
(iii-3) 反応溶媒の影響
反応機構を実験的に制御できる方法の1つに、溶媒の極性があります。水H2Oやアルコールはプロトン性極性溶媒ですが、これらの溶媒は、SN2およびSN1反応に、どのように影響を与えるでしょうか?溶媒の極性変化に伴うSN2反応の様子は、一見すると、複雑で分かりにくいです。溶媒の極性が増すに従って、ある場合は反応が速くなり、ある場合は反応が遅くなるからです。以下に2つの例を示します。最初の反応(a)は溶媒の極性が増すにつれて速くなりますが、2番目の反応(b)は遅くなります。
(a) (CH3CH2)3N + CH3CH2-I → [(CH3CH2)3Nδ+・・・ CH3CH2 ・・・ Iδ-] → (CH3CH2)3N+-CH2CH3 + I-
(b) HO- + CH3-S+(CH3)2 → [HOδ-・・・ CH3 ・・・ Sδ+(CH3)2] → HO-CH3 + S(CH3)2
この2種類のSN2反応の違いは、(a)では「生成物」で電荷の分離が起きていて、(b)では「反応物」で電荷の分離が起きていることです。これらの反応を、極性溶媒中で行うとどうなるでしょうか?極性の高い溶媒分子は、電荷を帯びた物質と相互作用をして、分子の形式電荷を非局在化させ、安定化させるはずです。つまり、(a)のような生成物で電荷の分離が起きている反応では、生成物が安定化され、(b)のような反応物で電荷の分離が起きている反応では、反応物が安定化されるのです。
反応速度は、遷移状態のエネルギー、すなわち活性化エネルギーによって決まります。(a)では、極性溶媒が生成物や電荷の分離が起き始めた遷移状態を安定化するので、活性化エネルギーが小さくなって、反応速度が速くなります。しかし、(b)では、極性溶媒が遷移状態や生成物よりもむしろ反応物を安定化してしまうので、活性化エネルギーが逆に大きくなって、反応速度が遅くなるのです。
また、SN1反応では、SN2反応と違って、反応溶媒の影響は、比較的単純です。SN1反応の最初の段階は、イオンの発生を伴う遅い反応段階です。極性溶媒はイオンを溶媒和できるので、その効果により、SN1反応は著しく促進されます。無極性溶媒では、このような効果はありません。SN1反応は、極性溶媒中では、反応速度が速くなるのです。
(iv) 求核置換反応のまとめ
求核置換反応には、SN2反応とSN1反応があり、どちらの反応が起こるのかを知ることによって、反応速度はどうであるのか、立体配置の反転が起こるのか、それともラセミ化が起こるのかなどということを、あらかじめ知ることができます。求核置換反応は、一方の反応が起こりにくい場合には、他方が有効であるというように、互いに相補的な関係です。例えば、SN2反応は、メチル化合物や第一級アルキル化合物でよく起こるのに対して、SN1反応は、第三級アルキル化合物でよく起こります。しかしながら、ちょうど中間に位置する第二級アルキル化合物では、両反応が起こりうると考えられます。R基が小さい場合には、強力な求核剤を用い、反応過程の電荷の様子に適した極性を持つ溶媒を選択することによって、SN2反応は進行しやすくなります。逆にR基がかさ高い場合には、小さい求核剤を用い、極性溶媒を用いることによって、SN1反応が進行しやすくなります。このように選択的に求核置換反応を起こすためには、それぞれの反応機構をよく知っておく必要があります。求核置換反応のまとめを、次の表.4に示します。
表.4 求核置換反応のまとめ
|
|
SN2反応 |
SN1反応 |
|
求核攻撃と脱離が同時 |
脱離 → 求核攻撃 |
|
|
R基 |
メチルまたは第一級アルキル化合物 |
第三級アルキル化合物 |
|
求核剤 |
優れた求核剤 |
あまり依存しない |
|
脱離基 |
優れた脱離基 |
優れた脱離基 |
|
反応溶媒 |
場合による |
極性溶媒 |
|
立体化学 |
ワルデン反転 |
ラセミ化 |
|
反応速度 |
反応基質と求核剤の濃度に比例 |
反応基質の濃度に比例 |
(3) 脱離反応(E2反応とE1反応)
有機化学では、他の分子を生成することなく、ある分子のみを100%の収率で与えるという反応は、ほとんどありません。有機合成化学の仕事の大部分は、選択性の探索ともいえます。合成化学者は、ある反応経路だけが、他のすべてに優先して進行するような反応試薬や反応条件を探索しています。求核置換反応を用いて、分子を合成しようとする場合には、特に「脱離反応(elimination reaction)」と呼ばれる副反応を考慮しなければなりません。例えば、ハロゲン化アルキルに求核剤を反応させると、置換反応と脱離反応の2つが、競争的に起こります。
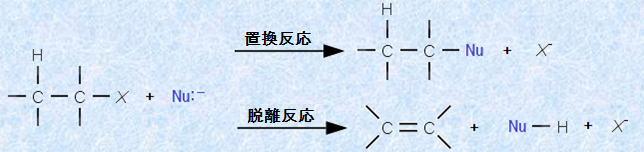
図.12 置換反応と脱離反応
置換反応では、ハロゲンが求核剤と置き換わります。一方で、脱離反応では、求核剤は塩基として働き、ハロゲンのある炭素の隣の炭素(α炭素)上からプロトンH+ を引き抜きます。その結果、ハロゲンと隣接炭素上の水素(α水素)とが脱離して、それらが結合していた炭素原子の間にπ結合が形成されるのです。このように、脱離反応は求核置換反応の副反応ではあるものの、ハロゲン化アルキルなどの基質からアルケンを得ることができるので、不飽和炭化水素の一般的な合成法として利用されています。
置換反応と脱離反応について、どちらの反応が主反応になるのかは、基質の構造や求核剤の種類、また反応条件などにより左右されます。ところで、この脱離反応にも、置換反応と同じように「E2」と「E1」の記号で表わす2つの主要な反応機構が存在します。この2つの反応を制御するためには、その反応機構をよく理解しておく必要があるでしょう。
(i) E2反応
「E2反応」は、SN2反応と同じように1段階反応です。ここでは、求核剤は塩基として働き、脱離基のある炭素に隣接した炭素(α炭素)上から、プロトンH+ を引き抜く働きをします。そして、それと同時に脱離基が離れ、二重結合が形成されます。この反応機構における電子対の動きを矢印で示すと、次の図.13のようになります。
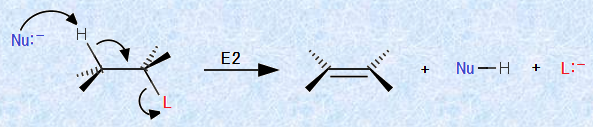
図.13 E2反応の反応機構
図.13では、E2反応が進行しやすい立体配座が書いてあります。すなわち、反応基質のH-C-C-Lとつながった4つの原子は、すべて1つの平面内にあり、C-HとC-L間の2面角は180°で、HとLは互いにアンチ型に配列しているということです。この反応では、C-HとC-L結合が長くなるに従い、アルケンのπ結合となる2p軌道が形成され始め、分子は平面構造に近付きます。したがって、新しくπ結合を形成するためには、炭素の2p軌道の方向が揃っていなければならず、C-HとC-Lの両結合が同時に切断できるアンチE2反応が、最も有利なのです。
E2反応は、SN2反応と競争的に起こりますが、生成物の割合は、第一に求核剤の塩基性に影響されます。強いブレンステッド塩基は、弱いブレンステッド塩基に比べて、プロトンH+ を引き抜くのに効果的です。したがって、塩基性の強い求核剤を用いると、E2反応が起こりやすく、塩基性の弱い求核剤を用いると、SN2反応が優先します。例えば、エトキシドイオンCH3CH2O- は優れた求核剤ですが、同時に塩基性も強いです。したがって、このような求核剤を反応で用いると、置換反応の副反応として、脱離反応が起こりやすくなります。置換反応を優先的に進めるためには、塩基性の弱い塩化物Cl- などを求核剤として用いる必要があります。
また、SN2反応の速度は、基質の構造に大きく影響されます。基質の枝分かれが多いほど、脱離基後方からの求核剤の攻撃に対する立体障害が大きくなるため、SN2反応の速度は減少するのです。先にも説明しましたが、第三級アルキル化合物では、SN2反応に対しての立体障害は極限に達し、SN2反応の速度は、実質的にゼロになります。SN2反応が遅くなるにつれて、これと競争して起こるE2反応が重要になってきます。臭化t -ブチルのような代表的な第三級アルキル化合物においては、E2とSN2反応の起こる条件下でも、E2反応のみが進行します。臭化t -ブチルのC-Br結合後方からの求核攻撃が立体的に起こりにくいのに対し、プロトンH+ の引き抜きは、さほど立体的な制約を受けないからです。
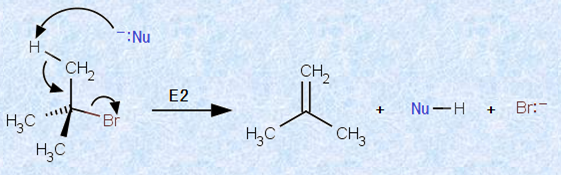
図.14 臭化t -ブチルのE2反応
(ii) E1反応
「E1反応」は、SN1反応と同じように2段階反応です。ここでは、求核剤は塩基として働き、脱離基のある炭素に隣接した炭素(α炭素)上から、プロトンH+ を引き抜く働きをします。反応の第1段階目では、反応基質がゆっくりと律速的にイオン化して、炭素陽イオンを生成します。そして、生じた炭素陽イオンから、プロトンH+ が引き抜かれて、二重結合が形成されます。この反応機構における電子対の動きを矢印で示すと、次の図.15のようになります。
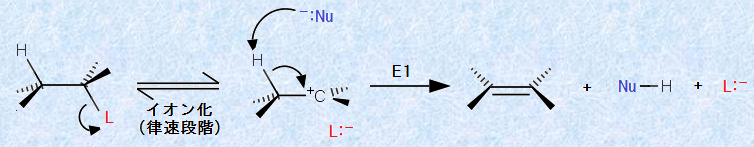
図.15 E1反応の反応機構
E1反応は、SN1反応と競争的に起こりますが、E1およびSN1生成物の割合は、求核剤の塩基性に左右されます。塩基性の強い求核剤は、プロトンH+ に対する親和性が高いので、SN1反応よりもE1反応に有利に働きます。例えば、同じ生成物を与える求核剤でも、エトキシドイオンCH3CH2O- では、塩基性が強すぎてE1反応が優先しますが、エタノールC2H5OHを求核剤として利用すれば、SN1反応が優先します。
(CH3)3C-Br + CH3CH2O- → (CH3)2C=CH2 + CH3CH2O-H + Br-
(CH3)3C-Br + CH3CH2OH → (CH3)3C-O-CH2CH3 + H-Br
(iii) ザイツェフ脱離
多くの脱離反応において、炭素陽イオン中間体から脱離しうる水素が、複数存在することがあります。脱離する水素によって生成物が異なる場合、主生成物は、より安定な多置換アルケンになります。多置換アルケンが安定な理由は、炭素二重結合に結合する置換基が多いほど、相対的にエネルギーの低いsp2-sp3結合が多くなり、置換基の少ないアルケンでは、相対的にエネルギーの高いsp3-sp3結合が多くなるからです。2s軌道にある電子は、2p軌道にある電子よりも、エネルギーが低く安定なので、軌道のs性が多くなるほど、その軌道にある電子の安定化は大きくなります。したがって、sp2混成軌道の炭素原子に結合している置換基の多いアルケンほど、その分子は安定になるのです。もちろん、統計学的確率も生成物の比に多少影響しますが、主たる要因はアルケンの安定性であり、脱離反応では、最も多くの置換基を持ったアルケンが、非常に優先して生成します。この現象は、ロシアの化学者であるアレクサンドル・ザイツェフによって発見されたので、「ザイツェフ脱離(Zaitzev elimination)」と呼ばれています。例えば、次の図.16のE1反応では、置換基の少ないアルケンの生成が統計的に有利であっても、より安定な多置換アルケンの生成が優先します。
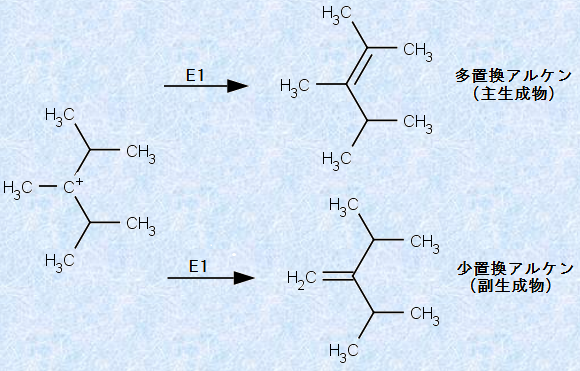
図.16 多置換アルケンを優先的に生成するザイツェフ脱離
・参考文献
1) H.ハート/L.E.クレーン/D.J.ハート 共著「ハート基礎有機化学」培風館(1986年発行)
2) メートランド・ジョーンズ「ジョーンズ有機化学(上)」東京化学同人(2000年発行)