�E�L�@�����@�\(�F�������d�q�u������)
�y�ڎ��z
(i) �F�������d�q�u�������̔����@�\
(1) �F������
�u�F����������(aromatic compounds)�v�́A�x���[��C6H6���\�Ƃ����s�O�a�L�@�������̈�Q�ł��B���ɒY�����f�����ō\���������̂��A�u�F�����Y�����f(aromatic hydrocarbon)�v�Ƃ����܂��B���q���Ƀx���[���̍\�������������̉��������A���L�̖F���������Ƃ���u�F����(aromatic)�v�Ɩ��t����ꂽ�̂ł����A�K�������x���[���̍\���������������u�F��(aroma)�v�����Ƃ͌���܂���B���݂́A���̓��F���鉻�w�����Ƃ��̉��w�I���萫��\���Ӗ��Ƃ��āA�u�F�����v�Ƃ������t���g���Ă��܂��B���Ȃ킿�A���q���Ƀx���[���̍\�������������͔M�͊w�I�Ɉ���ł���A���̓��ʂȈ��萫����ʓI�Ɂu�F������(aromaticity)�v�Ƃ����܂��B�Ȃ��F�����������́A���ٓI�Ɉ���Ȃ̂ł��傤���H
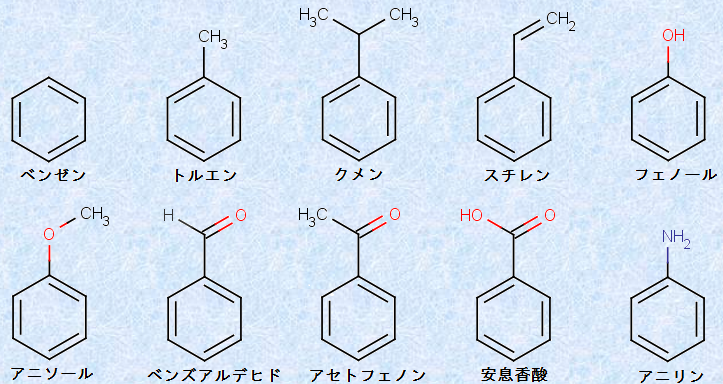
�}.1 �l�X�ȖF����������
�}.1�Ɏ����悤�ɁA�F�������ƌĂ����ʂ̈��萫��L���鉻�������A�x���[���ȊO�ɂ��������݂��Ă��܂��B�����ŁA���̂悤�ȉ��������`�t�����ʑ������o�����߂ɁA�܂��͌��^�ƂȂ�x���[���̍\���I�������܂Ƃ߂Ă݂܂��傤�B
���ɁA�x���[���́u��\���v�����܂��B���������āA�F�������́A�������������ł���Ƃ����܂��B�����đ��ɁA�x���[���́u���S����(fully conjugated)�v���Ă��܂��B���S�����Ƃ́A�x���[���̊�̊e�Y�f��2p�O����1�������A�אڂ���2p�O�������ׂČq�����Ă����Ԃ��w���܂��B�Ⴆ�A���̐}.2�Ɏ���1,3,5-�V�N���w�v�^�g���G�������Ă݂�ƁA�̒[��2p�O���̏d�Ȃ荇�����r��Ă���̂ŁA���̂悤�ȉ������͖F�������������܂���B
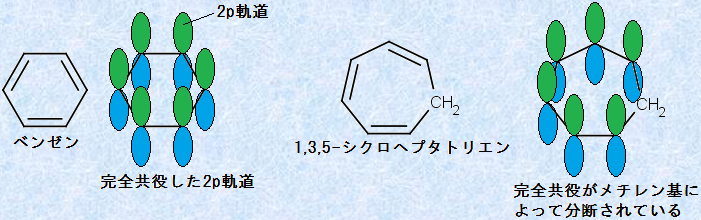
�}.2 �F�������������x���[���ƁA�F�������������Ȃ�1,3,5-�V�N���w�v�^�g���G��
��O�ɁA�x���[���́u���ʍ\���v�������܂��B���̕��ʐ��ɂ��A2p�O���̏d�Ȃ荇�����ő�ƂȂ�̂ł��B�����x���[���̊\�������ʂłȂ��Ȃ�A2p�O���̏d�Ȃ荇�����\���łȂ��A2p�O���Ԃ̏d�Ȃ荇���ɂ�蓾������萫���A�����������Ă��܂��܂��B���ʍ\������傫�������ƁA2p�O�����m�̊�̏d�Ȃ荇�����A�قƂ�ǐ�Ă��܂��̂ł��B���̗ǂ��Ⴊ�A���̐}.3�Ɏ����V�N���I�N�^�e�g���G���ł��B���̕��q�͗����̂悤�Ȍ`�����Ă���A2p�O�����������Ă��邽�߂ɁA2p�O���Ԃ̏d�Ȃ荇�����قƂ�ǂ���܂���B
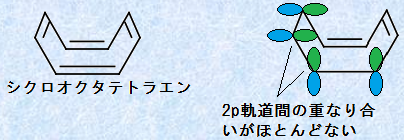
�}.3 ���ʕ��q�łȂ��V�N���I�N�^�e�g���G��
����ł́A��̕��ʍ\���������A���S�������Ă��邷�ׂĂ̕��q�́A�x���[���̂悤�ȖF����������萫�����̂ł��傤���H�����́A���炩�ɔۂł��B�Ⴆ�A�V�N���u�^�W�G���́A����3�̋K�������ׂĖ������Ă��܂����A�|200���Ƃ����ቷ�ł��������Ă��܂��قǁA�ɂ߂ĕs����ȕ��q�ł��邱�Ƃ��m���Ă��܂��B�V�N���u�^�W�G���́A�����V���v���ȍ\���Ȃ���A���o�����Ƃ͑�ς�����A20���I�㔼�ɂ́u�L�@���w�̐��t�v�Ƃ����Ă�A�����̉Ȋw�҂��������������������ƂŒm���Ă��܂��B�V�N���u�^�W�G���́A�ʍ\���Ŋ��S�����ł���Ȃ���A�F��������S�������Ȃ�������(���F������)�ł��B
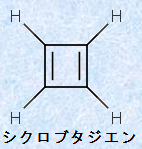
�}.4 �ʍ\���Ŋ��S�����ł���Ȃ���A���ɕs����ȃV�N���u�^�W�G��
�܂�A�F�������̏����Ƃ��ẮA�܂����������Ă���̂ł��B���̏�����������ɂ́A�x���[���ƃV�N���u�^�W�G���̈Ⴂ���l����K�v������܂��B���̍ŏI�I�Ȉ�ʑ����A1931�N�Ƀh�C�c�̕������w�҂ł���G�[���q�E�q���b�P���ɂ���Ē�Ă���܂����B�q���b�P���́A�x���[�����q�S�̂����d�q�̋O�����ߎ��I�ɎZ�o������@��҂ݏo�����̂ł��B���̌��_�����������Ă��܂��A�u�𐬂��Ă������d�q�̐���4n�{2(n��0,1,2,������)�̂Ƃ��A���̕��q�͖F�������������Ĉ��艻����v�Ƃ������ƂɂȂ�܂��B������u�q���b�P����(Huckel�fs rule)�v�Ƃ����܂��B�x���[���́A�܂��ɂ���n��1�̏ꍇ�ɓ��Ă͂܂邽�߁A���Ɉ���ȕ��q�Ƃ��đ��݂ł���̂ł��B�Ȃ��An�͊��̌��q���Ƃ́A�S���W���Ȃ����Ƃɒ��ӂ��ĉ������B���̌��q���͊W�Ȃ��ɁA���d�q�̐��������l����Ηǂ��̂ł��B
|
�q���b�P����(Huckel�fs rule) |
|
�@ ��\���ł��� �A �A������2p�O���������A���S�������Ă��� �B ���ʍ\���ł��� �C 4n�{2�����d�q������ |
�q���b�P������K�p����ƁA�x���[���͊����d�q��6���̂ŁAn��1�̖F�����������ł���A�����d�q��10���i�t�^������n��2�A�����d�q��14���A���g���Z����n��3�̖F�����������ł��B�����̕��q�́A�q���b�P�����������ɖ������Ă��邱�Ƃ�������܂��B
�}.5 �F�����������́A���d�q��4n�{2����
�������Ȃ���A�Ȃ����d�q�̐����d�v�Ȃ̂ł��傤���H�q���b�P�����́u4n�{2�v�́A��̂ǂ̂悤�ȈӖ������̂ł��傤���H����͊ȒP�Ɍ����A���ʊ�\���������A���S��������4n�{2�����d�q�������q�݂̂��A�x���[���Ɠ��l�́u���q�O��(molecular orbital)�v��������ł��B�܂�A����������ɂ���u���������q�O��(bonding molecular orbital)�v�ɁA���ׂĂ����d�q�����܂�A�s���艻�Ɋ�^����u�����������q�O��(antibonding molecular orbital)�v�ɂ��A���艻��s���艻�Ɋ֗^���Ȃ��u�������q�O��(nonbonding molecular orbital)�v�ɂ��A���d�q���S������Ȃ��̂��A4n�{2�����d�q�����ꍇ�Ȃ̂ł��B���̐}.6�ɁA�x���[���������q�O���������܂��B�x���[����6�����d�q�O�������ݍ�p����ƁA6�̕��q�O�����ł�������܂����A���̋O���́A3�̌������O����3�̔��������O���ł��B
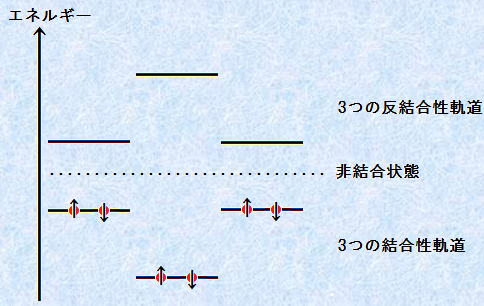
�}.6 �x���[���������q�O��
�x���[���ɂ����ẮA�������̕��q�O���͍ő���g���Ă���A��������߂铭�������锽�������O���ɂ́A�S���d�q�������Ă��܂���B����̂ɁA�x���[���͔��Ɉ���ȍ\���Ȃ̂ł��B���l�ɖF�������������i�t�^�����ł́A���������q�O���́A���̐}.7�̂悤�ɂȂ�܂��B�i�t�^������10�̊����d�q�������A�����̓x���[���Ɠ����悤�ɁA���ׂĈ���Ȍ������O���ɓ���܂��B�܂��A�}.7�Ŏ����悤�ɁA�F�������������Ȃ��V�N���u�^�W�G���ɂ́A4�̊����d�q������A���̂���2�͈���Ȍ������O���Ɏ��܂�܂����A�c���2�͔����O���ɓ���܂��B�����O���́A���q�̈��艻�ɂ͊�^���Ȃ��̂ŁA�V�N���u�^�W�G���̐����́A�x���[���Ƃ͑S���قȂ��Ă��܂��B����ǂ��납�A�V�N���u�^�W�G���͌����Ԃ̓��p��90���ł��邱�Ƃ���A�傫�Șc�݂������Ă���A�ނ���s����ȉ������ł��B
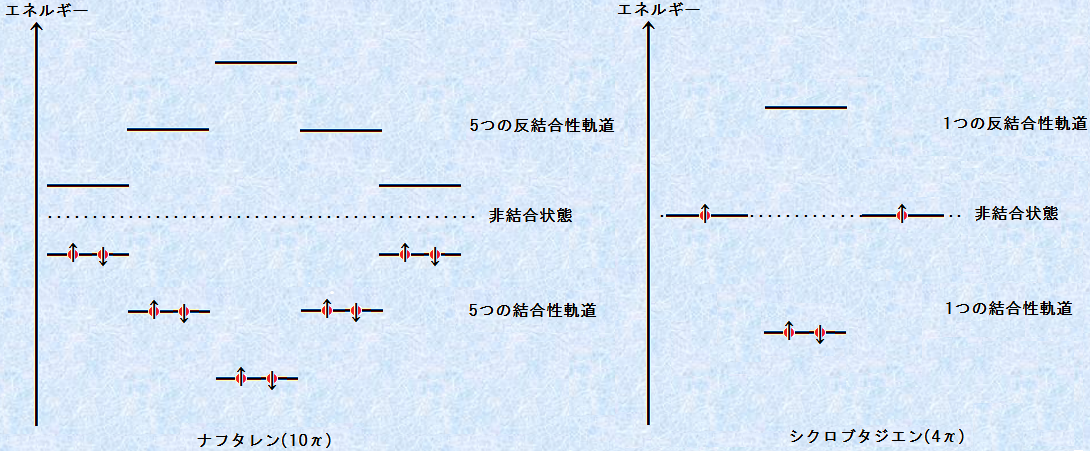
�}.7 �i�t�^�����ƃV�N���u�^�W�G���������q�O��
(2) �F�������d�q�u������
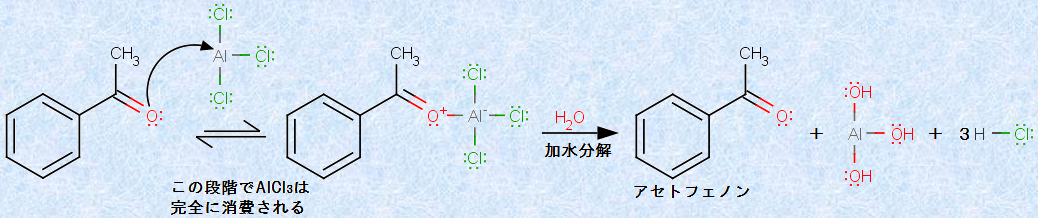
�F�����������̔������́A����ȖF�������́u6���d�q�n�v��ۂƂ��Ƃ��鐫���Ɏx�z����Ă��܂��B���̖F��������j��ɂ́A���ɑ傫�ȃG�l���M�[��K�v�Ƃ��A�t�ɖF�����������߂����Ƃ́A���ɗe�ՂȂ��Ƃł��B�F�����������̍ł���ʓI�Ȕ����́A�F����̐��f���A���̌��q��u����Œu��������^�C�v�̂��̂ł��B�x���[���̑�\�I�ȁu�u������(substitution reaction)�v�̂��������A���̐}.8�Ɏ����܂��B
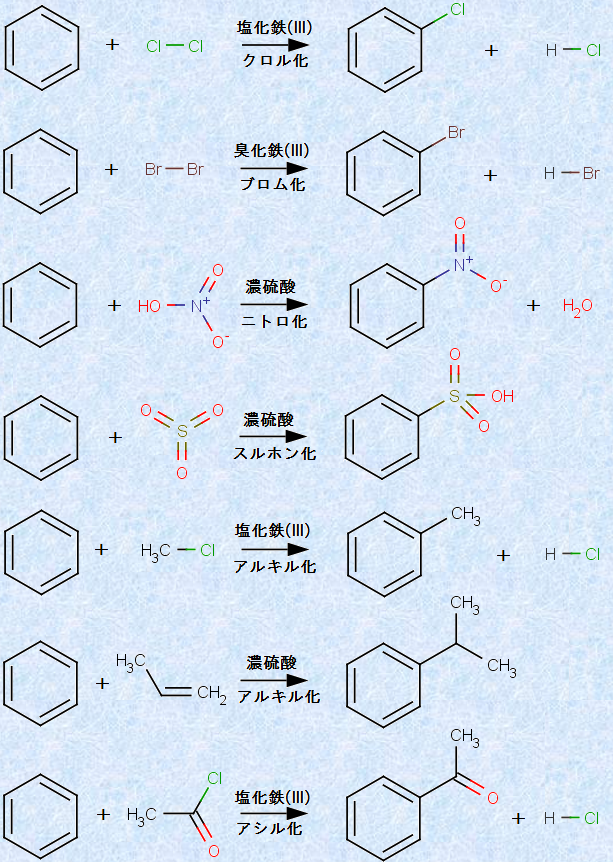
�}.8 �x���[���̑�\�I�Ȓu������
�����̔����̂قƂ�ǂ́A0�`50���̔������x�ŏ����ɐi�s���܂��B�������A�x���[����ɂ��炩���ߒu������݂���Ƃ��́A�u����ɉ����āA�������������₩�ɂ�����A���邢�͉ߍ��ɒ��߂����肵�Ȃ���Ȃ�܂���B�܂��A�����u�����2�ȏ㓱������Ƃ��ɂ��A����������ύX���Ȃ���Ȃ�܂���B�����̔����̋N�������A�t�����������u���������N����₷�����R�A�G�}�̖����Ȃǂɂ��ẮA���ŏq�ׂ܂��B
(i) �F�������d�q�u�������̔����@�\
�}.8�Ŏ����������̂��ׂĂ��A�x���[���ɑ��鋭�͂ȁu���d�q��(electrophilic reagent)�v�̍U���Ŏn�܂邱�Ƃ́A�����̏؋��ɂ�蕪�����Ă��܂��B���͂ȋ��d�q�܂́A�x���[�������d�q�_����^���������d�q2���g���ăx���[���ɕt�����A�x���[����1�̒Y�f���q�����������`�����܂��B���̌��ʁA���̒Y�f���q��sp2��������sp3�����ƂȂ�A����ȖF������������6���d�q�n�͔j���̂ŁA�x���[���̈��艻�G�l���M�[�͎����Ă��܂��܂��B���̂悤�ɁA���d�q�܂��x���[���ɕt�����āA6���d�q�n��j�邽�߂ɂ́A���ꂾ���̃G�l���M�[�Ƌ��͂ȋ��d�q�܂��K�v�ƂȂ�̂ł��B�܂��A�����ł̓x���[�����A���d�q�܂ɑ���u���d�q���^��(���C�X����)�v�Ƃ��ē����Ă��邱�ƂɂȂ�܂��B
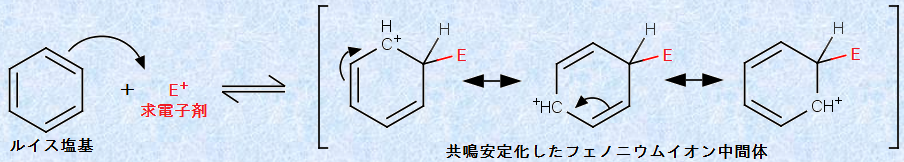
�}.9 �x���[���ɑ��鋁�d�q�܂̕t��
���̔����̌��ʐ�����Y�f�z�C�I�����A�u�t�F�m�j�E���C�I��(phenonium ion)�v�ƌĂ�ł��܂��B���̒Y�f�z�C�I���̐��d�ׂ́A���d�q�܂��t������sp3�Y�f���猩��o -�����p -�̈ʒu�ɁA���ɂ���Ǎ݉����邱�Ƃ��ł��܂��B�t�F�m�j�E���C�I���͋��ɂ����艻���Ă��܂����A���̗z�C�I�����������邱�Ƃɂ�莸����F�������̈��艻�G�l���M�[�́A�}.9�̂悤�ȋ����艻�ɂ���Ă��A�ق�̈ꕔ�������߂��܂���B�����ŁA�x���[�m�j�E���C�I���́A���d�q�܂��t�������Y�f���q��̐��f���qH���v���g��H+ �Ƃ��ĕ��o���邱�Ƃɂ��A6���d�q�n�̃x���[�����Đ������āA���������������܂��B
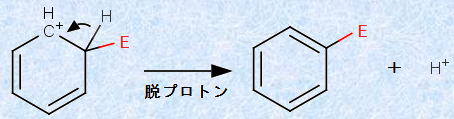
�}.10 �x���[���̍Đ�
�}.9�Ɏ��������d�q�܂̕t���́A�F�������̔j��ɂ��Ȃ�̊������G�l���M�[���K�v�ƂȂ�܂��B���̂��߁A���̒i�K�̔������x�͒ʏ�x���A�����������́u�����i�K(rate-limiting step)�v�ƂȂ�܂��B�}.10�Ɏ������x���[���̍Đ��́A6���d�q�n���Đ������̂ŁA�Ⴂ�������G�l���M�[�������ƂɂȂ�A�������x�͒ʏ푬���ł��B�����̔����́A�u�����鋁�d�q�܂��������āA��ʓI�Ɂu�F�������d�q�u������(electrophilic aromatic substitution)�v�ƌĂ�܂��B
(ii) �n���Q����
�P���ȃA���P���Ƃ͈قȂ�A�x���[���́A�L�fBr2�≖�fCl2�Ƃ͖������Ŕ������܂���B�u�n���Q��������(halogenation reaction)�v���N�������߂ɂ́A�ʏ�A�L���S(III) FeBr3�≖���S(III) FeCl3�Ȃǂ̂悤�ȁA�Ή������n���Q�����S�G�}���K�v�ƂȂ�܂��B���̗��R�́A���fCl2�́A���C�X����̎ア�x���[���Ɣ�������قǂ́A���͂ȋ��d�q��(���C�X�_)�ł͂Ȃ�����ł��B�����ŁA�S�G�}�́A���fCl2��F�����������Ɣ������鋭�͂ȋ��d�q�܂ɕς��铭�������܂��B���Ȃ킿�A�����S(III) FeCl3�́A���fCl2�ƍ��̂��`�����邱�ƂŁA���fCl2������������̂ł��B
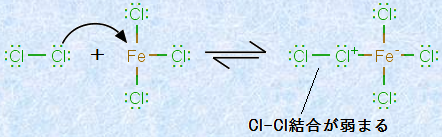
�}.11 �����S(III) FeCl3�Ɖ��fCl2�̍��̌`��
�x���[���́ACl2-FeCl3���̗̂D�ꂽ�E����ł���FeCl4- �ƒu������̂ɁA�\���ȃ��C�X����������܂��B���̒u���ɂ��A�t�F�m�j�E���C�I�����ԑ̂������āA�����ăv���g��H+ ���E�����A�N�����x���[�����������܂�(���f���Fchlorination)�B���̔����́A������Ɍ����邩������܂��A�A���R�[���̃q�h���L�V��(-OH)���v���g�������āA�D�ꂽ�E����ł���+OH2��ɕς��锽���Ɏ��Ă��܂��B�����S(III) FeCl3�́A�A���R�[������������ۂ̃v���g��H+ �Ɠ��l�̖��������Ă���Ƃ����܂��B�L�fBr2�ƏL���S(III) FeBr3��p����u�L�f��(bromination)�v�̔����@�\�������ł��B
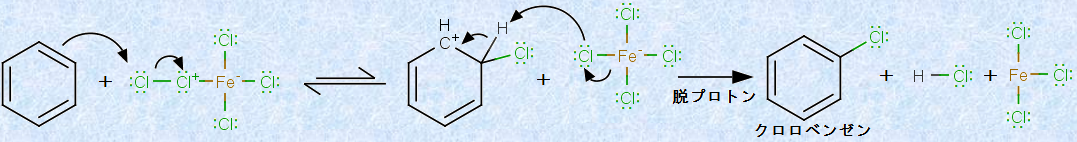
�}.12 �x���[���̉��f��
(iii) �j�g����
�j�g����(-NO2)��F���ɓ������邽�߂ɂ́A���_�_���������ŏɎ_HNO3����p�����A�F�������d�q�u���������N�����܂��B���_H2SO4(Ka��108)�́A�Ɏ_HNO3(Ka��102)��苭���_�ł��邩��A���_���ł͏Ɏ_HNO3�̓v���g��������AH2O-NO2+ ���܂��B�����āA���ꂩ�琅H2O������ƁA���͂ȃ��C�X�_�ł���j�g���j�E���C�I��NO2+ ���������A���ꂪ�F�������d�q�u�������ɂ����鋁�d�q�܂Ƃ��ē����̂ł��B���̔����́A��ʓI�Ɂu�j�g����(nitration)�v�ƌĂ�܂��B
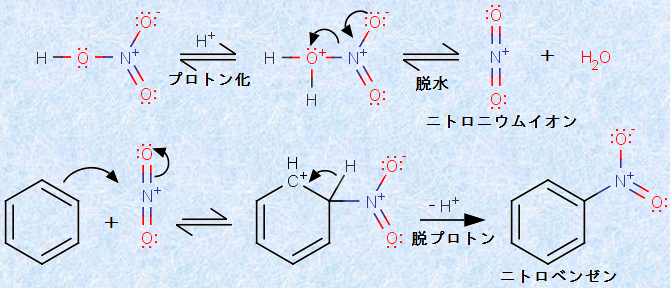
�}.13 �x���[���̃j�g����
(iv) �X���z����
�@�X���z��(-SO3H)��F���ɓ������邽�߂ɂ́A�Z���_H2SO4���邢�͔Z���_�ɉߏ�̎O�_������SO3���z���������������_���g�p����܂��B�����Ŕ����ɂ������鋁�d�q�܂́A�O�_������SO3�܂��̓v���g�������ꂽ�O�_������SO3H+ �ł��BSO3�͋��d�q�����\���ɍ����̂ŁA�v���g��������Ȃ��Ă��A�x���[���Ɣ������܂��B���̔����́A��ʓI�Ɂu�X���z����(sulfonation)�v�ƌĂ�A�������̃X���z���_�́A�����L�@�_�ł��B
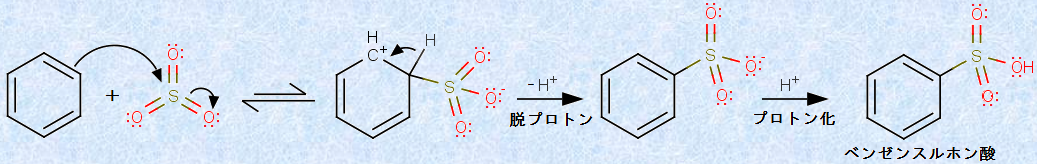
�}.14 �x���[���̃X���z����
(v) �A���L����
�F�����������́u�A���L����(alkylation)�v�́A��ʓI�Ɂu�t���[�f���E�N���t�c����(Friedel-Crafts reaction)�v�ƌĂ�܂��B���̖��̂́A�t�����X�̉��w�҂ł���V�������E�t���[�f���ƁA�A�����J�̉��w�҂ł���W�F�[���Y�E�N���t�c��2�l���A1877�N�ɏ��߂Ă��̔��������o�������Ƃɂ��Ȃ�ŁA���t�����Ă��܂��B���̔����̋��d�q�܂́A�Y�f�z�C�I���ł���A(a)�n���Q�����A���L���Ƀ��C�X�_�G�}(�����A���~�j�E��AlCl3�Ȃ�)�����āA�n���Q�������C�I����E����������@��A(b)�A���P���Ƀv���g��H+ ��t����������@�ɂ�蔭�������܂��B���̐}.15�ɁA�G�`���x���[���̍������ɂƂ��āA���̔����o�H���ڂ��������Ă����܂��傤�B
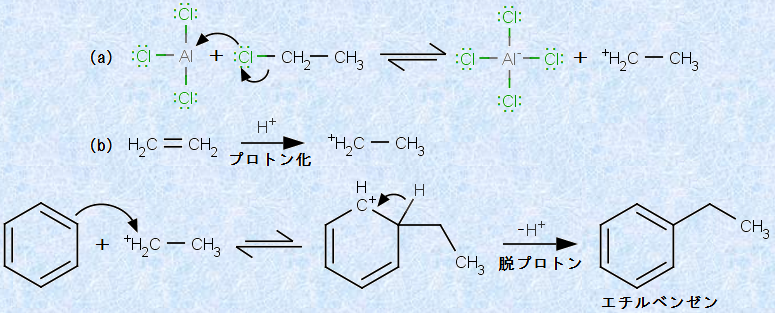
�}.15 �x���[���̃A���L����
�������A�t���[�f���E�N���t�c�A���L���������̓K�p�ɂ́A���x������܂��B���̔����́A���ʃj�g����(-NO2)��X���z��i-SO3H�j�Ȃǂ��x���[����ɑ��݂���ꍇ�ɂ́A�p���邱�Ƃ��ł��܂���B���̗��R�́A�����̒u����A�G�}�̉����A���~�j�E��AlCl3�ƍ��̂��`������ƁA������������������ł��B
(vi) �A�V����
�t���[�f���E�N���t�c�A���L���������̕ό`�ł���u�t���[�f���E�N���t�c�A�V�����v�ɂ���āA�A�V����(-COR)��F���ɓ������邱�Ƃ��ł��܂��B�����A�V���́A�����A���~�j�E��AlCl3���݉��ŁA�x���[���Ɣ������܂��B���̏ꍇ�A�u�G�}�ʁv�ł͂Ȃ��A�u�������ʁv�̉����A���~�j�E��AlCl3���K�v�ƂȂ�܂��B�����A�V���́A�����A���~�j�E��AlCl3�ƍ��̂��`�����A�����艻���ꂽ�A�V���E���C�I�������܂��B���̎��ɁA�A�V���E���C�I���̓x���[���ɕt�����A�t�F�m�j�E���C�I�����ԑ̂����܂��B�����āA���C�X����v���g��H+ ��E��������Ɣ����͊������A�������̃A�V���x���[����^���܂��B��Ƃ��āA���̐}.16�ɁA�A�Z�g�t�F�m���̍��������������܂��B
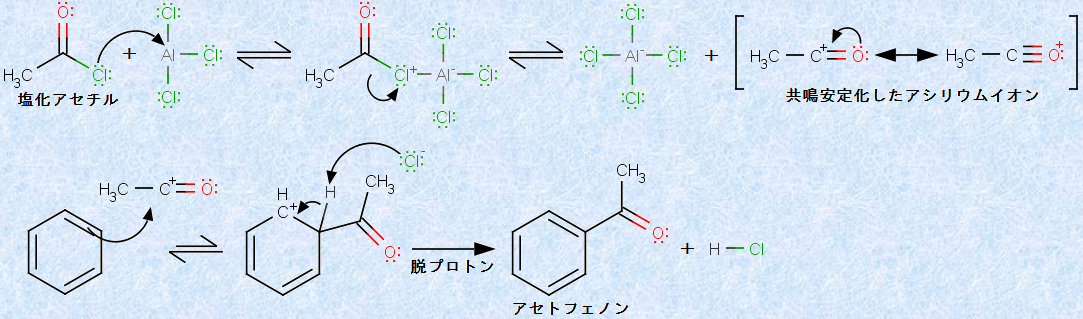
�}.16 �x���[���̃A�V����
��ɂ��q�ׂ��悤�ɁA�t���[�f���E�N���t�c�A���L���������ƈقȂ�A�t���[�f���E�N���t�c�A�V���������́A�G�}�����ł͂���܂���B�������̃A�V���x���[�����A�����A���~�j�E��AlCl3�ɑ��Ĕ�����������̂ŁA��������₢�Ȃ�A1�F1���̂��`������̂ł��B�܂�A�����Ő��������A�V���x���[���Ɠ��ʂ̉����A���~�j�E��AlCl3���A���̌`���Ŏg���܂��B���������āA���������������邽�߂ɂ́A���w�ʘ_�ʂ̉����A���~�j�E��AlCl3���K�v�ƂȂ�܂��B�����I����ɐ�H2O��������ƁA���͕̂�������A�A�V���x���[�����V�����܂��B�����@�\�����G�ł���ɂ�������炸�A�t���[�f���E�N���t�c�A�V���������͗L�p�Ȕ����ł���A��X�̖F�����P�g�����A�����悭�������܂��B
�}.17 ���������ɂ��A�A�V���x���[�����V������
(3) �z�����ʂƊ���������
�u��u���x���[���v�́A���d�q�܂Ɣ�������ƁA��ʓI�ɂǂ̂悤�Ȑ�������^����ł��傤���H�܂��A�������ɂ́A�ǂ̂悤�ȉ\��������ł��傤���H�u��u���x���[���v�ɂ́A�u�I���g(ortho�F1,2-��u��)�́v��u���^(meta�F1,3-��u��)�́v�A�u�p��(para�F1,4-��u��)�́v��3��ނِ̈��̂����݂��܂��B
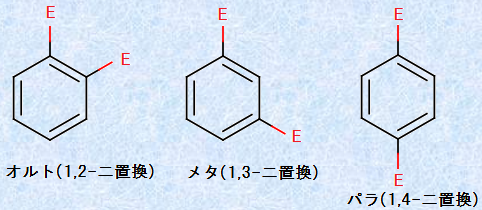
�}.18 3��ނ̓�u���x���[��
�x���[����̒u����̎�ނɂ���āA���������u���x���[���ِ̈��̔䂪�A�傫���ς��܂��B��u���x���[���̒u���ɂ���Đ��������u���x���[���́A�I���g�F���^�F�p���ِ̈��̔�́A2�F2�F1�̓��v�I�ȕ��z�ɂ͂Ȃ�܂���B�����̈�u���x���[���̔������ׂ�ƁA�u�I���g��u���̂ƃp����u���̂���ɗ^�������(�I���g-�p���z����)�v�ƁA�u���^��u���̂�D�悵�ė^�������(���^�z����)�v�ɕ��ނ���܂��B
�܂��A�V���������Ă���u����̈ʒu�Ƃ��̔������x�ɂ��A���ւ�����܂��B���Ȃ킿�A�u�I���g�́v�Ɓu�p���́v����ɗ^�����u���x���[���́A�x���[�����g�ɔ�ׂĔ������x�������ł��B�t�ɁA�u���^�́v��^�����u���x���[���́A�x���[�����g�ɔ�ׂĔ������x���x���ł��B
�\.1 �u���x���[���̔z�����ʂƊ���������
|
|
�u����̎�� |
�u����̖��� |
���������x |
|
|
H(�x���[��) |
|
1 |
|
�I���g-�p���z���� |
-NH2�A-NHR�A-NR2 |
�A�~�m�� |
���ɑ��� |
|
-OH |
�q�h���L�V�� |
���ɑ��� |
|
|
-OR |
�A���R�L�V�� |
���ɑ��� |
|
|
-R |
�A���L���� |
���� |
|
|
-NHCOR |
�A�V���A�~�m�� |
���� |
|
|
-F-�A-Cl�A-Br�A-I |
�n���Q�m�� |
�x�� |
|
|
���^�z���� |
-COR |
�A�V���� |
�x�� |
|
-COOH |
�J���{�L�V�� |
�x�� |
|
|
-CONH2 |
�A�~�h |
�x�� |
|
|
-COOR |
�G�X�e�� |
�x�� |
|
|
-SO3H |
�X���z�� |
�x�� |
|
|
-C��N |
�V�A�m�� |
�x�� |
|
|
-NO2 |
�j�g���� |
�x�� |
�x���[������ɂ��Ĕ�ׂ�ƁA�q�h���L�V��(-OH)��`����(-CH3)�̂悤�Ȓu����́A�F���̔��������x���[���������߂܂����A�N������(-Cl)��j�g����(-NO2)�̂悤�Ȓu����́A�t�ɔ�������ቺ�����܂��B���`����(-CH3)�́A���f(-H)�ɔ�ׂēd�q���^�I�ł���A�N������(-Cl)��j�g����(-NO2)�́A���f(-H)�ɔ�ׂēd�q�����I�ł��邱�Ƃ́A���łɐ������̏؋����番�����Ă��邱�Ƃł��B��ʓI�ɓd�q���^���̒u����́A�F��������d�q���^�\�͂����߂܂��B����ŁA�d�q�������̒u����́A�F��������d�q���^�\�͂�ቺ�����܂��B
�u���x���[���̔����́A�F�������d�q�u�������̋@�\�Ɗ��S�Ɉ�v���Ă��܂��B���Ȃ킿�A�������x���F���ւ̋��d�q�܂̍U���i�K�Ō��܂�̂Ȃ�A��ɑ��݂���d�q���^���̒u����͔������������A���ɓd�q�������̒u����͔�����x������͂��ł��B�u����̐����ɂ���āA�F���̔��������A���̂悤��2�ʂ�̉e�����邱�Ƃ́A�\.1�Ŏ������悤�ɁA���ׂĂ̖F�����u�������ł��ϑ�����Ă��܂��B
(i) �I���g-�p���z����
�g���G�����j�g��������ƁA�uo -�j�g���g���G���v�Ȃ�тɁup -�j�g���g���G���v����ɐ������܂����A�um -�j�g���g���G���v�͂قƂ�ǐ������܂���B���̂悤�ɁA�g���G���̃��`����(-CH3)�́A�u�I���g-�p���z�����v�̒u����ł��B���̗��R���A���d�q�F�����u�������̔����@�\��K�p���āA�������܂��傤�B�����@�\�̏��߂̒i�K�ŁA�j�g���j�E���C�I�����A���`����(-CH3)�ɑ��āAo -, m -, p -�̊e�ʒu���U�������Ƃ��̗l�q���A���̐}.19�Ɏ����܂��B
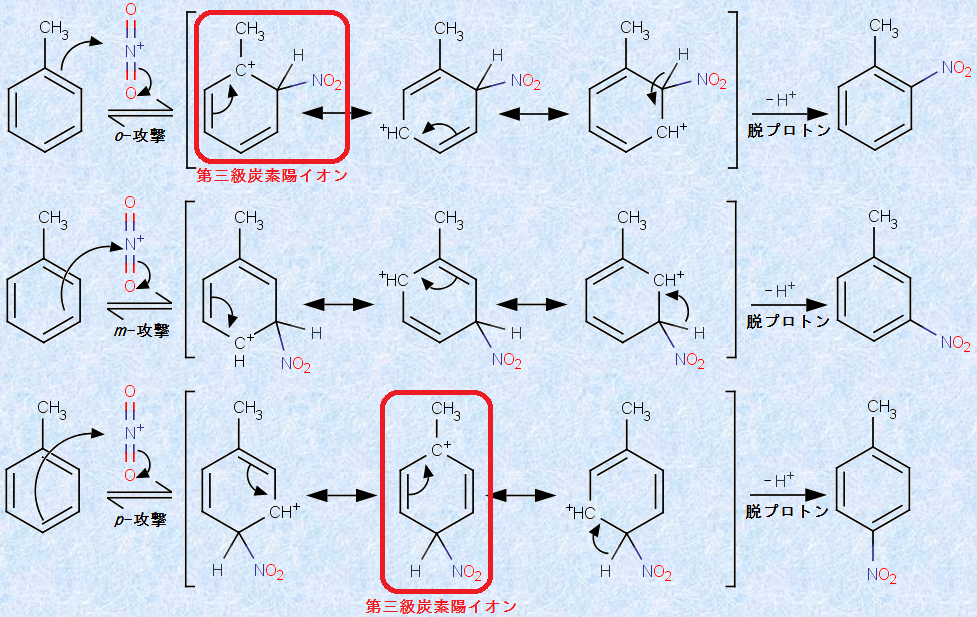
�}.19 �g���G���̃I���g-�p���z����
�����ŁA�������ԑ̂ł���t�F�m�j�E���C�I���̐��d�ׂ�тт��Y�f�ɒ��ڂ���ƁAm -�U���Ő����锽�����ԑ̂̐��d�ׂ́A���ׂđ�Y�f��ɑ��݂��܂��B����ŁAo -�U����p -�U���Ő����锽�����ԑ̂ł́A���d�ׂ�тт�3�̒Y�f�̂���1�͑�O���Y�f�ł���A���d�ׂ̈��艻�ɓ��ɓK���Ă��܂�(�}.19���Ԙg�̍\��)�B��O���Y�f�z�C�I���́A�ł�����ȒY�f�z�C�I���ł���A���̔����́A�ł�����ȒY�f�z�C�I�����ԑ̂��o�R���Đi�s���܂��B���̌��ʂƂ��āA���`����(-CH3)�́A�u�I���g-�p���z�����v�������̂ł��B�܂��A����Ɠ��l�ɁA���̃A���L������A���ׂăI���g-�p���z�����������܂��B
�@����ɁA�\.1�Ŏ������A���̃I���g-�p���z�����u����ɂ��Ă��l���Ă݂܂��傤�B�A�~�m��(-NH)�A�q�h���L�V��(-OH)�A�A���R�L�V��(-OR)�A�A�~�h(-NHCOR)�A�N������(-Cl)�Ȃǂ̒u����́A���ׂĖF���ɒ����������q��ɔL�d�q�������Ă���̂ŁA���ꂪ�����邱�Ƃɂ���āA�אڈʂ̐��d�ׂ����艻�ł��܂��B�����ł͗�Ƃ��āA�t�F�m�[���̃��`���������グ�āA�l�@���Ă݂܂��傤�B
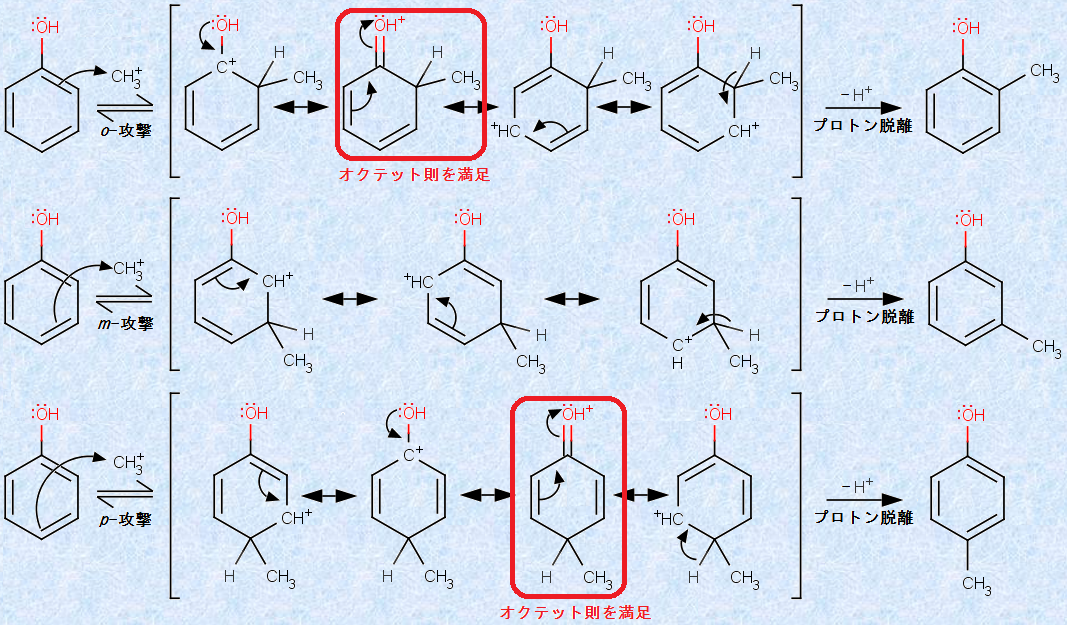
�}.20 �t�F�m�[���̃I���g-�p���z����
o -�U����p -�U���Ő�����t�F�m�j�E�����ԑ̂̋��\����1���A�q�h���L�V��(-OH)�̌��������Y�f��ɐ��d�ׂ𑶍݂����Ă��܂��B�����ŁA���̍\���̎_�f���q�̎��L�d�q���A�אڂ��鐳�d�ׂ��������Y�f���q��Ɉړ�������ƁA���d�ׂ��_�f���q�Ɉړ������\�����������Ƃ��ł��܂�(�}.20�Ԙg�̍\��)�B���̍\���́A�I�N�e�b�g�������\���ł���A����6�d�q��̋��\���ɔ�ׂāA���Ɉ��艻����Ă��܂��B���̂悤�ȃI�N�b�e�g�������\���́Am -�U���ɂ����ď������Ƃ��ł��܂���B���̌��ʂƂ��āA�q�h���L�V��(-OH)�́A�u�I���g-�p���z�����v�������̂ł��B�����ŏq�ׂ��l�@�͈�ʐ��������Ă���A�F���ɒ����������q��ɔL�d�q�������Ă���u����́A���ׂăI���g-�p���z�����ł��B
(ii) ���^�z����
�j�g���x���[���̃j�g���������ɂ��Ă��A�j�g����(-NO2)�́um -�z�����ʁv���A�����悤�ɐ����ł���ł��傤���H�j�g���x���[���ł́A���f���q��Ɍ`���d�ׁ{1�����݂���̂ŁA������l���ɓ���āA�������N���邻�ꂼ��̈ʒu�ɂ��āA�t�F�m�j�E���C�I�����ԑ̂������Ă݂�ƁA���̐}.21�̂悤�ɂȂ�܂��B
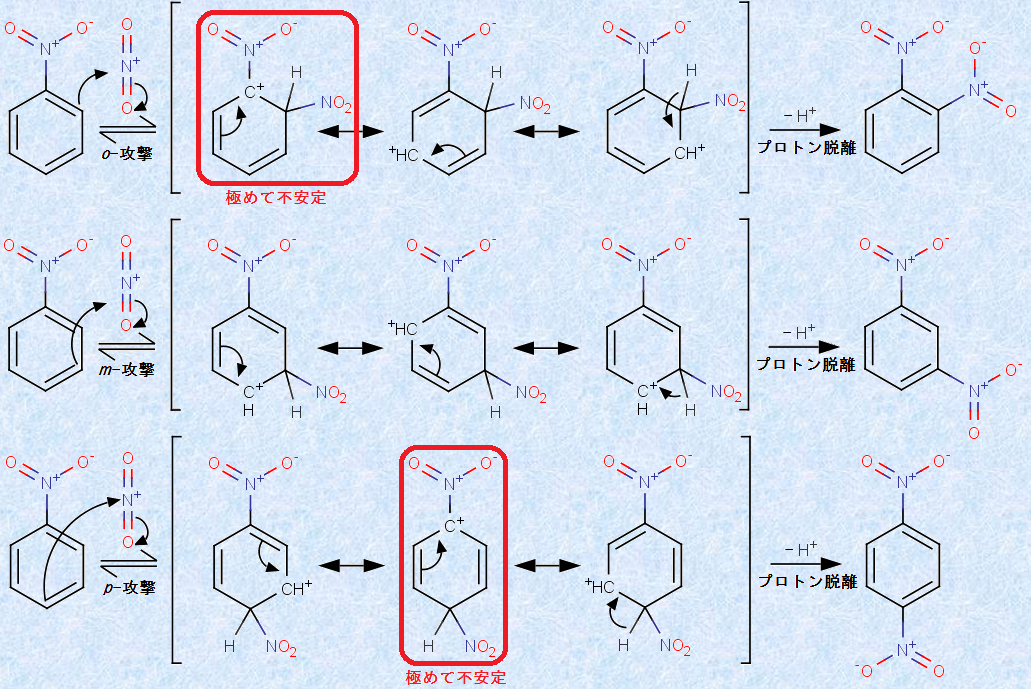
�}.21 �j�g���x���[���̃��^�z����
o -�U���Ȃ�т�p -�U���Ő�����t�F�m�j�E�����ԑ̂ɂ��ď����ꂽ���\�����̂����A�Ԙg�ň͂����\���́A2�̐��d�ׂ�ׂ荇�킹�Ɏ����Ă���A�����d�ד��m���������������߂ɁA�ɂ߂čD�܂����Ȃ��z�u�ł��B����ɑ��āAm -�U���Ő�����t�F�m�j�E���C�I�����ԑ̂́A���̂悤�ȍD�܂����Ȃ����\�������������Ƃ��ł��Ȃ��̂ŁA���ʂƂ��āAo -�U���Ȃ�т�p -�U���Ɣ�r���āAm -�U�����L���ɂȂ��ł��B
����Ɠ����������A�\.1�ɋL�ڂ������ׂĂ̊��\��ɓ��Ă͂߂邱�Ƃ��ł��܂��B����烁�^�z�����u����߂ċC�t�����Ƃ́A�F���ɒ������錴�q���A�s�O�a�������\�����錴�q��1�ł���A����ɗאڂ��錴�q�́A�傫�ȓd�C�A���x��������(�Ⴆ��O��N)�ł���Ƃ������Ƃł��B�����̒u����ł́A�j�g����(-NO2)�̒��f���q�̂悤�ɁA�F���ɒ����������q�͕����I�ɐ��d�ׂ������Ă���Ao -�U���Ȃ�т�p -�U���Ő�����t�F�m�j�E���C�I�����ԑ̂̍\�����������Ƃ��ɁA���d�ׂ��ׂ荇���s����ȍ\���������邽�߂ɁA���ʂƂ��āu���^�z�����v�������̂ł��B
�E�Q�l����
1) ���������Y�u���������q ���E�͘Z�p�`�łł��Ă���v�u�k��(2019�N���s)
2) H.�n�[�g/L.E.�N���[��/D.J.�n�[�g �����u�n�[�g��b�L�@���w�v�|����(1986�N���s)
3) ���[�g�����h�E�W���[���Y�u�W���[���Y�L�@���w(��)�v�������w���l(2000�N���s)