・有機反応機構(カルボン酸とその誘導体の反応)
【目次】
(1) カルボン酸の誘導体
カルボン酸誘導体は、カルボキシ基(carboxy group, -COOH)のヒドロキシ基(-OH)部分を、他の置換基Lで置換した化合物です。酸ハロゲン化物や酸無水物、エステル、アミドは、すべてカルボン酸誘導体です。これらは、一般式RCOLを持つ「アシル化合物(acyl compound)」と呼ばれています。これらのアシル化合物は加水分解をすると、すべて原料のカルボン酸に戻ることができます。次の図.1に、カルボン酸とその誘導体の一般的な構造式を示します。
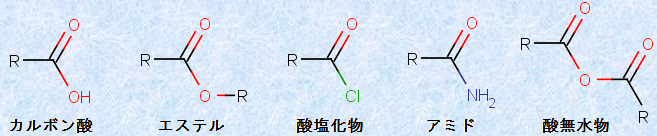
図.1 カルボン酸とその誘導体の一般的な構造式
これらの分子は、構造的に似ているだけでなく、化学反応も密接に関連しており、あるアシル化合物の反応は、一般的に他のアシル化合物の合成に相当します。幸いにも、これら見かけ上ずいぶん異なる化合物の間でも、共通する性質があるのです。アシル化合物の構造は、主に置換基Lとカルボニル基(-CO-)の相互作用によって決まり、その化学変化では、置換基Lが他の置換基に置き換わる置換反応が一般的です。次の図.2に、様々なアシル基(acyl group, RCO-)を示します。
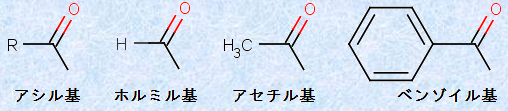
図.2 様々なアシル基(RCO-)
(2) フィッシャーのエステル化反応
「エステル(ester)」は、カルボン酸の誘導体であり、ヒドロキシ基(-OH)の水素がRに置き換えられています。カルボン酸とアルコールを、濃硫酸H2SO4などの酸触媒存在下で加熱すると、エステルと水H2Oが生成し、原料との間に平衡反応が成立します。
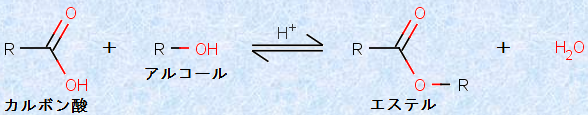
図.3 フィッシャーのエステル化反応
この反応方法は、ドイツの有機化学者であるエミール・フィッシャーが開発したものであり、その名にちなんで、「フィッシャーのエステル化反応(Fischer esterification)」と呼ばれています。この反応は平衡反応ですが、この平衡を右側に移動させる方法は、いくつかあります。例えば、アルコールかカルボン酸のいずれか値段の安い方を大過剰に用いるとか、生成するエステルや水H2Oを蒸留などにより反応系から取り除けば、ル・シャトリエの法則により、反応は右側へ進行します。
フィッシャーのエステル化反応を考えてみると、生成物の水分子が、「酸のOHとアルコールのHから生成するのか」それとも「酸のHとアルコールのOHから生成するのか」という、反応機構上の疑問が浮かんできます。この疑問は、取るに足らない小さなことのように思うかもしれませんが、実はこれを解き明かすことで、エステルや他のアシル化合物の化学的性質を理解することができるのです。
RCOOH + ROH ⇄ RCOOR + H2O
RCOOH + ROH ⇄ RCOOR + H2O
この反応の反応機構を書くことは、実はそれほど難しくありません。というのも、その重要な段階は、他のカルボニル基(-CO-)を含む化合物の反応によく似ているからです。反応の第一段階は、アルデヒドやケトンの反応と同じく、カルボン酸のカルボニル酸素のプロトン化です。このとき、共鳴安定化された中間体が生成します。
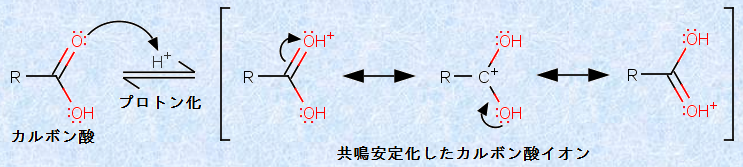
図.4 カルボニル酸素のプロトン化(第一段階)
反応の第二段階は、やはりアルデヒドやケトンの化学に類似しており、アルコール分子が、プロトン化されたカルボニル基(-CO-)に求核付加します。プロトン化によって、カルボキシ基(-COOH)の炭素原子上の正電荷が増加し、炭素原子上への求核攻撃が起こりやすくなっているのです。この段階で、新たな炭素-酸素結合(エステル結合)が形成されます。
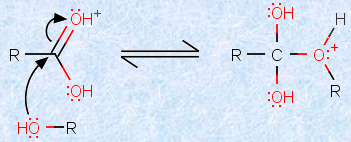
図.5 アルコール分子の求核付加(第二段階)
反応の第三段階および第四段階は、ルイス塩基性の酸素にプロトンが付加したり、脱離したりする平衡反応です。この酸塩基平衡は、可逆的で速く、酸素を含む化合物の酸性溶液中では、常に起こる平衡です。第四段階での2つのヒドロキシ基(-OH)は等価であり、どちらにプロトン化が起こっても、同じ結果になります。
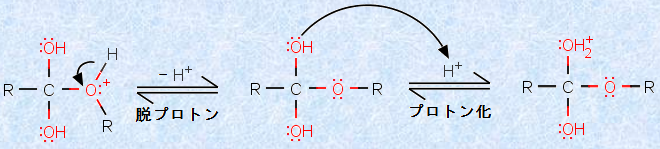
図.6 プロトン化と脱プロトン(第三段階および第四段階)
反応の第五段階では、炭素-酸素結合が開裂して、水分子が脱離します。脱離基については、その塩基性が弱いほど脱離能が良いので、水分子は比較的優れた脱離基です。ヒドロキシ基(-OH)のままでは、塩基性が強すぎて脱離しにくいので、第4段階でプロトン化して、脱離しやすい水H2Oを作っているのです。
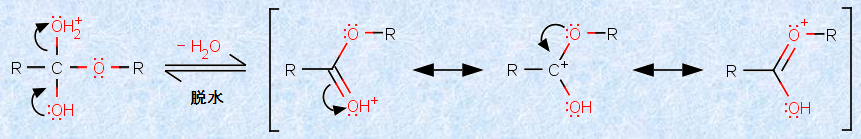
図.7 水分子の脱離(第五段階)
反応の第六段階では、脱プロトンによってエステルが生成し、酸触媒が再生します。ただし、これらの反応はすべて平衡反応であり、基本的には、エステルからカルボン酸に戻る加水分解反応も、並行して存在していることに注意が必要です。
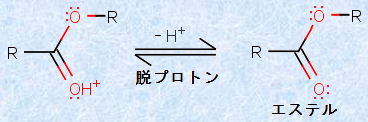
図.8 エステルの生成(第六段階)
アルデヒドやケトンの反応性は、カルボン酸やその誘導体のアシル化合物と、どのように違うのでしょうか?これはすなわち、アルデヒドやケトンには脱離する基がなく、カルボン酸やその誘導体には脱離する基が存在することです。したがって、この反応は特に、「求核的アシル置換反応(nucleophilic acyl substitution)」と呼ばれています。しかし、この反応は、実際には1段階の置換反応ではなく、求核付加に続いて脱離が起こる2段階反応であることに注意が必要です。
フィッシャーのエステル化反応は基本的な反応であり、この反応をよく知れば、細部が異なるとしても、多くの類型的な反応に一般的な応用ができます。このような意味で、フィッシャーのエステル化反応は、十分に理解しておくべき反応です。
(3) 求核的アシル置換反応
カルボン酸やエステル、およびその関連化合物のほとんどの反応は、カルボニル炭素上への求核攻撃から始まります。例えば、フィッシャーのエステル化反応では、最初にアルコール分子が、プロトン化されたカルボニル基(-CO-)を求核攻撃します。そして、カルボニル基(-CO-)の再成とともに脱離基が脱離すれば、反応は完了します。このように、すべての求核的アシル置換反応は、付加-脱離過程による反応をするのです。次の図.9に、求核的アシル置換反応の一般的な反応機構を示します。これは、アシル基(RCO-)を持つすべての化合物に応用ができます。
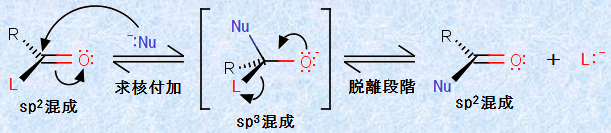
図.9 求核的アシル置換反応
この反応は、結果的に求核剤Nuと脱離基Lの置換が起こるので、「アシル基移動反応(acyl transfer reaction)」とも呼ばれています。反応の第一段階では、平面三方形であったカルボニル炭素が、求核剤Nuの攻撃を受けて、四面体形の中間体を形成します。つまり、炭素原子の混成が、sp2混成からsp3混成へと変化するのです。そして、その次に脱離基Lが外れて、平面三方形の炭素原子を持ったカルボニル基(-CO-)が再成します。この結果、アシル基(RCO-)に置換している脱離基Lを、求核剤Nuで置換したことになるのです。
求核的アシル置換反応では、脱離基Lの性質が、反応段階の速度に影響を及ぼします。すなわち、脱離基Lの電子吸引性(電気陰性度)が増大すると、求核的アシル置換反応における2つの段階の反応速度は増大します。これは、反応の第一段階では、Lの電気陰性度が大きいほど、カルボニル炭素がより正の電荷を帯びるために、求核攻撃を受けやすくなるからです。さらに、反応の第二段階でも、Lの電気陰性度が大きくなると、脱離基の負電荷が安定化して脱離能が高まるので、各段階の反応が進みやすくなります。
また、一般的にエステルは、アルデヒドやケトンよりも、求核剤に対する反応性が低いです。この理由は、エステルでは、カルボニル炭素上の正電荷が、酸素原子上に非局在化できるからです。一般的には、その物質における共鳴構造式が多く書けるほど、その物質は安定になります、それ故に、エステルは、アルデヒドやケトンよりも、求核剤に対する反応性が低くなります。
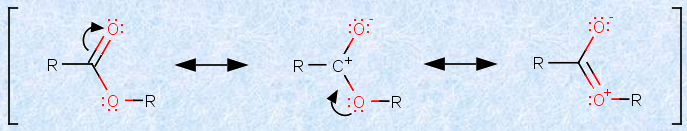
図.10 エステルの共鳴安定化は、カルボニル炭素の正電荷密度を低くする
(i) 酸ハロゲン化物の反応
すべてのアシル化合物は、付加-脱離過程による反応をします。「酸ハロゲン化物(acid halide)」は、アシル化合物の中でも、特に求核剤に対して反応性が高いです。これは、ハロゲンの電気陰性度が大きいために、カルボニル炭素の部分正電荷密度が高くなり、またハロゲン物イオンは、一般的に優れた脱離基であるからです。酸ハロゲン化物の中で、最も一般的で廉価なものは、酸塩化物です。そのため、非常に多くのアシル化合物が、酸塩化物を出発物質として、合成されています。
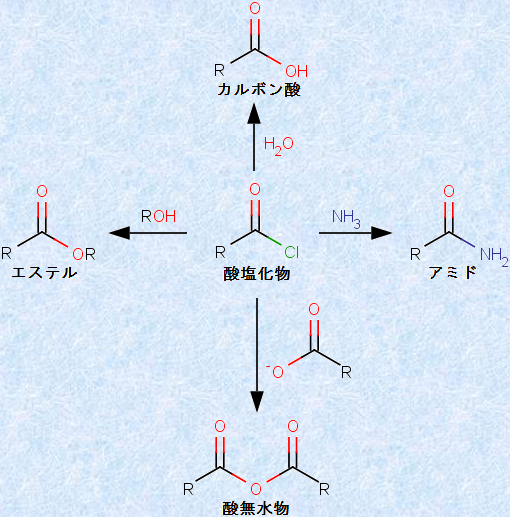
図.11 酸塩化物を用いたアシル化合物の合成
ハロゲン化アシルは、ほとんどの求核剤と速やかに反応します。例えば、塩化アセチルは、湿気と急速に反応して加水分解されます。このため、酸ハロゲン化物には、一般的に刺激臭があり、塩化ベンゾイルなどには、催涙性があります。
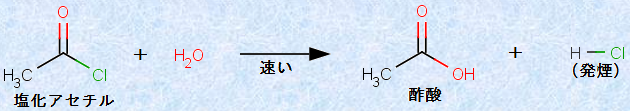
図.12 塩化アセチルの加水分解
ハロゲン化アシルは、アルコールとも速やかに反応して、エステルを生成します。例えば、塩化ベンゾイルは、メタノールCH3OHと室温で反応して、安息香酸メチルを与えます。
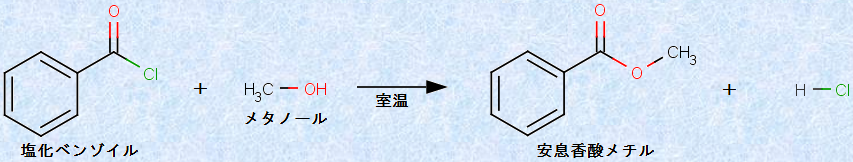
図.13 塩化ベンゾイルとメタノールCH3OHの反応
実験室でエステルを合成するときに用いられる一般的な方法は、ハロゲン化アシルをアルコールと反応させる方法です。フィッシャーのエステル化反応は、非常に有名な反応ですが、この反応は平衡反応であるために、反応原料のどちらか1つを、通常大過剰に用いる必要があったことを思い出して下さい。ハロゲン化アシルを用いる方法は、原料のカルボン酸またはアルコールが、高価である場合に有用な反応です。
さらに、ハロゲン化アシルは、アンモニアNH3とも速やかに反応して、アミドを生成します。この反応は有用ですが、発生する塩化水素HClを中和しなければならないので、アンモニアNH3が合計2当量必要であることに注意して下さい。

図.14 塩化アセチルとアンモニアNH3の反応
(ii) 酸無水物の反応
「酸無水物(acid anhydride)」は、2つのカルボキシ基(-COOH)から水分子を取り去って、互いに結合させたものです。脂肪族の無水物のうち、最も代表的なものは、無水酢酸(CH3CO)2Oです。年間約100万tが生産され、主にアルコールと反応させて、酢酸エステルを製造するのに使用されています。無水酢酸(CH3CO)2Oの最もよく知られた用途は、アスピリン(アセチルサリチル酸)の合成です。この反応では、サリチル酸のフェノール性ヒドロキシ基(-OH)がアセチル化されます。アスピリンは、消炎・解熱・鎮痛作用を持ち、高校化学の教科書にも登場する代表的な医薬品の1つです。
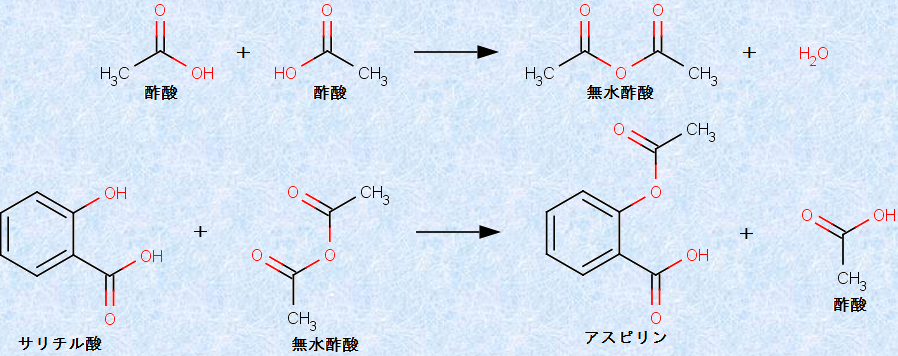
図.15 無水酢酸(CH3CO)2Oを用いたアスピリンの合成
酸無水物の反応は、定性的には酸ハロゲン化物に似ています。カルボン酸イオンが、付加-脱離過程における脱離基として働き、様々なアシル化合物が合成できます。酸無水物の求核剤に対する反応性は、一般的にエステルよりも高いですが、酸ハロゲン化物よりは低いです。この理由は、これらのアシル化合物から生じる脱離基の塩基性が、RO->RCOO->X- であることから説明できます。一般的に塩基性が弱いほど、脱離能は高くなるので、脱離能は逆にX->RCOO->RO- となり、反応性も同じ順になるのです。無水酢酸(CH3CO)2Oの代表的な求核的アシル置換反応を、次の図.16に示します。
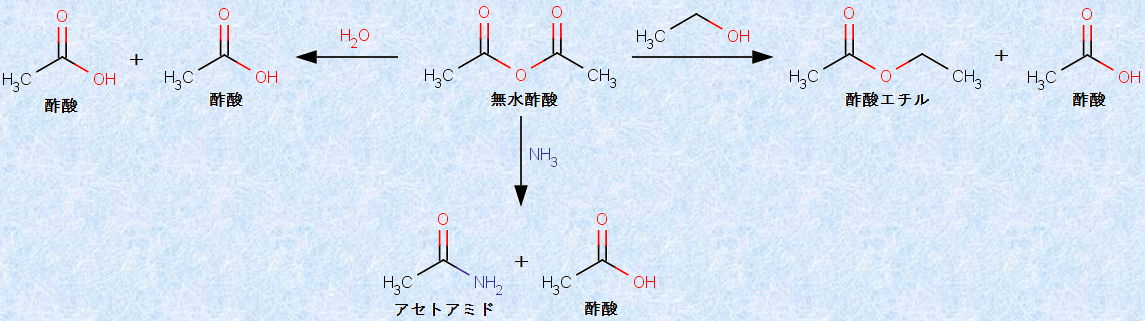
図.16 無水酢酸(CH3CO)2Oの代表的な求核的アシル置換反応
無水酢酸(CH3CO)2Oを加水分解すると、カルボン酸である酢酸CH3COOHに戻ります。無水酢酸(CH3CO)2Oをアルコールと反応させるとエステルになり、またアンモニアNH3との反応ではアミドになり、いずれの場合も酢酸CH3COOHが1当量生成してきます。
(iii) アミドの反応
「アミド(amide)」は、一般的なカルボン酸誘導体の中では、最も反応性が低いです。その結果、この官能基を持った化合物は、自然界に広く存在しています。最も重要なアミドはタンパク質です。タンパク質におけるアミド結合は、特に「ペプチド結合(peptide bond)」と呼ばれています。一般式RCONH2で表される第一級アミドは、アンモニアNH3をエステルや酸ハロゲン化物、酸無水物などと反応させて合成されます。また、アミドは、カルボン酸のアンモニウム塩を加熱することによっても得られます。
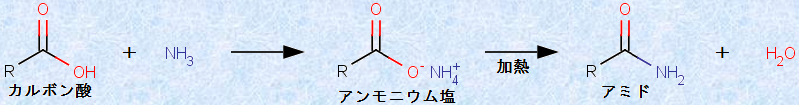
図.17 カルボン酸を用いたアミドの合成
アミドは、平面構造を持っています。炭素-窒素結合は、一般的に単結合として書かれますが、実際には、この結合軸まわりの回転は、二重結合のようにかなり束縛されています。その理由は、アミドにおける次の図.18のような共鳴の寄与が、極めて重要だからです。
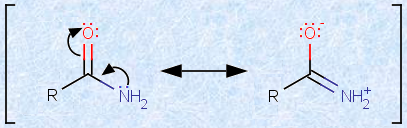
図.18 アミドの共鳴
この炭素-窒素二重結合を持つ構造の寄与が大きいので、実際の炭素-窒素結合は、かなり二重結合に似た挙動を示します。その結果、アミド結合のCとNに結合する2個ずつの原子は、すべて同一平面に存在することになり、炭素-窒素結合の結合軸まわりの回転は、かなり束縛されます。実際に、アミド結合の炭素-窒素結合の長さは0.132 nmしかなく、一般的な炭素-窒素単結合(約0.147 nm)よりもかなり短いです。そして、この共鳴安定化の寄与が大きいため、アミドは他のアシル化合物よりも安定になり、反応性が低くなるのです。
しかしながら、他のアシル化合物と同じように、アミドは求核剤と反応することができます。例えば、アセトアミドは、次の図.19で示すように、水H2Oと反応して加水分解されます。ただし、アミドは安定であるため、その反応速度は遅く、通常は長時間の加熱と、酸や塩基のような触媒を必要とします。
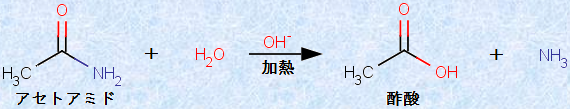
図.19 アセトアミドの加水分解
(iv) 求核的アシル置換反応のまとめ
求核的アシル置換反応は、次の表.1のようにまとめることができます。表.1の左側には、求核剤に対する反応性の減少順に、4つのアシル化合物が示されています。上の行には、代表的な求核剤が3つ示されています。注目するべきことは、それぞれの縦の欄に示された生成物が、左側に示された原料のアシル化合物の種類を問わず、同じものになることです。
表.1 求核的アシル置換反応のまとめ
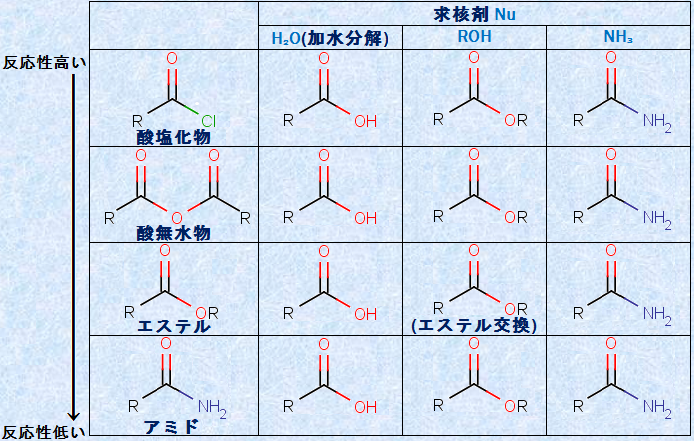
例えば、原料に酸塩化物や酸無水物、エステル、アミドのいずれを用いても、加水分解反応の生成物は、同じカルボン酸です。同様に、アルコールとの反応生成物は同じエステルであり、アンモニアNH3との反応生成物は同じアミドです。そして、表.1のすべての反応は、図.9で示したように、求核剤がアシル化合物のカルボニル基(-CO-)を攻撃することにより開始されます。この考えを応用すれば、アシル化合物と様々な求核剤との反応生成物が予想できます。
(4) クライゼン縮合
エステルのα水素は、アルデヒドやケトンほど酸性が強くありません。そのpKa値は、およそ24であることが分かっています。つまり、エステルはケトンなどより、pKa値で約5単位ほど弱い酸なのです。この理由は、エステルの場合、アルコキシ基(-OR)の酸素原子が、カルボニル基(-CO-)に対して電子供与的に作用して、α炭素上の負電荷が非局在化しにくくなるからです。
しかし、アルコキシドイオンのような強塩基が溶液中に存在すれば、わずかに脱プロトンして、エノラートアニオンが生成します。エステルより生じたエノラートアニオンは、「エステルエノラート(ester enolate)」と呼ばれます。
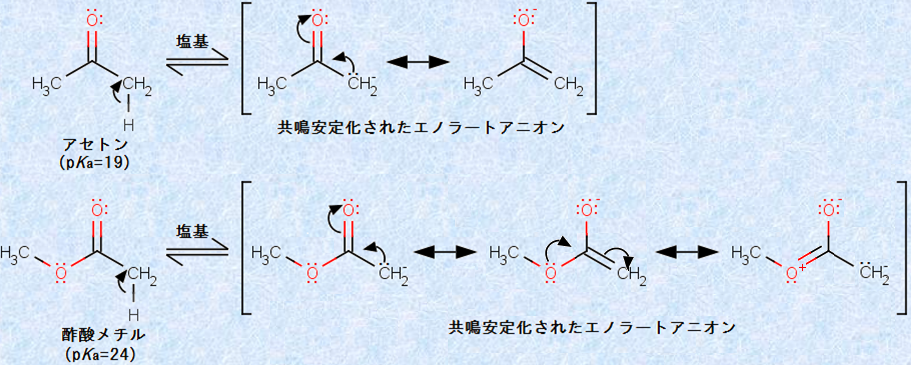
図.20 エステルとケトンのエノラートアニオン
しかしながら、アルコキシドイオンがカルボニル基(-CO-)に付加すると、塩基触媒によるエステル交換反応が起こるのではないでしょうか?実は、この反応が起こるため、アルコキシドイオンOR- とエステルのOR基を同じにしなければ、エステルの混合物が生成してしまいます。しかし、ORの構造が同じである限り、塩基触媒によるエステル交換反応が起こっても、生成物は出発物質と同じになり、見かけ上は何の変化も起こりません。
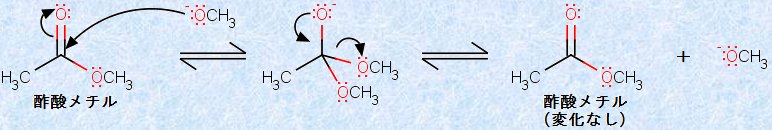
図.21 エステル交換反応
エステルエノラートは、どのように反応するでしょうか?エステルエノラートは、生成すると炭素型の求核剤として働くので、もう1分子のエステルのカルボニル基(-CO-)に求核付加を起こします。この反応は、「クライゼン縮合(Claisen condensation)」と呼ばれ、β -ケトエステルの合成法として利用されています。
例として、次の図.22に、酢酸エチルの「クライゼン縮合」を示します。生成物は、代表的なβ -ケトエステルであるアセト酢酸エチルです。反応の第一段階では、炭素型の求核剤であるエステルエノラートが、エステルのカルボニル基(-CO-)に求核付加します。続いて、反応の第二段階では、カルボニル基(-CO-)の再成とともにエトキシドイオンが脱離し、β -ケトエステルが生成するのです。この反応は、四面体の中間体を経由して進行し、これらの第一段階や第二段階の反応は、いずれも完全に可逆的な反応です。
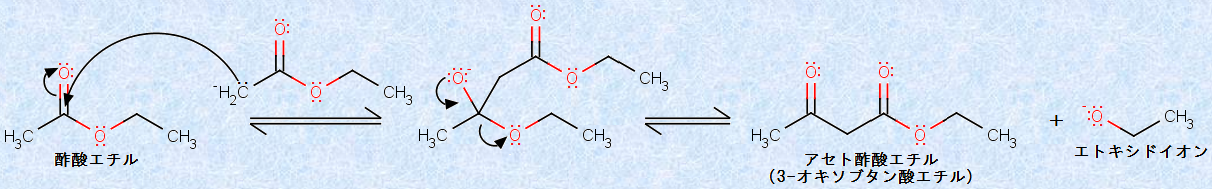
図.22 酢酸エチルのクライゼン縮合
ケトンやアルデヒドで起こるアルドール反応のように、クライゼン縮合においても、熱力学的に有利なのは出発物質です。エステルは、共鳴によって安定化されており、出発系には2個のエステル結合があるのに対して、生成系には1個しかありません。その結果、2個の別々のエステルの方が、熱力学的には有利なのです。
しかし、実際には、この熱力学的に不利な状況は、次のようにして解消されています。クライゼン縮合の最終段階では、触媒であるアルコキシドイオンが再生されています。このアルコキシドイオンは、生成物のβ -ケトエステルをエノラートアニオンに変換し、生成系を大きく安定化して、第一段階と第二段階の平衡反応を、生成物側に進行させる働きを持つのです。次の図.23に、アセト酢酸エチルのエノラートアニオンを示します。
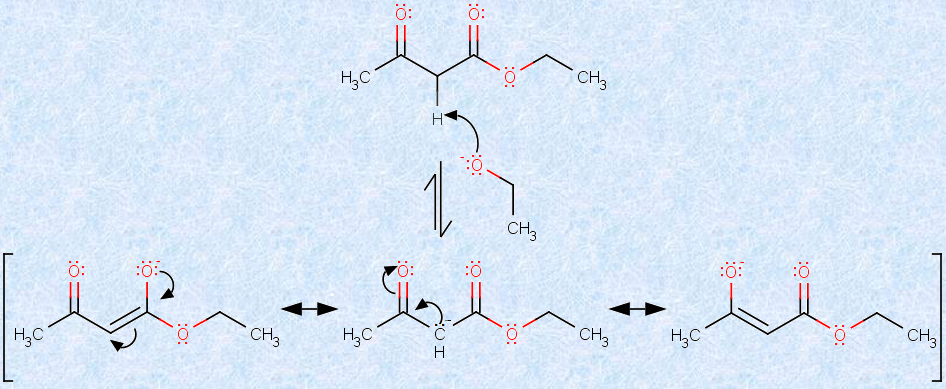
図.23 アセト酢酸エチルのエノラートアニオンの共鳴構造式
アセト酢酸エチルのメチレン水素CH2は、2つのカルボニル基(-CO-)に挟まれており、通常のエステルのα位の水素と比べて、はるかに強い酸性を示します(pKa=12)。クライゼン縮合の第一段階と第二段階の平衡反応は吸熱的ですが、この最終段階の酸塩基反応は発熱的なので、結果として、平衡が生成物側に移動することになるのです。
しかし、最終的に酸塩基反応が起こると、反応の触媒は消費されてしまいます。したがって、もし「触媒量」のアルコキシドイオンでクライゼン縮合を試みても、それは失敗します。用いたアルコキシドイオンと同量の生成物しか得られません。「触媒量」ではなく、「等モル量」のアルコキシドイオンを用いる必要があるのです。つまり、熱力学的に有利なクライゼン縮合は触媒的反応ではなく、等モル量の塩基を必要とするのです。
アルドール反応では、塩基が触媒量で良かったのに、クライゼン縮合では、塩基が化学量論量必要である理由をしっかり把握して下さい。そして、このクライゼン縮合を終了させるには、反応溶液を酸性にします。反応溶液に酸を加えると、生じたエノラートアニオンにプロトン化が起こり、β -ケトエステルが生成します。

図.24 クライゼン縮合後のプロトン化
・参考文献
1) H.ハート/L.E.クレーン/D.J.ハート 共著「ハート基礎有機化学」培風館(1986年発行)
2) メートランド・ジョーンズ「ジョーンズ有機化学(下)」東京化学同人(2000年発行)