�E�m�l�q(�j���C����)�����@�̊�b
�y�ڎ��z
(5) �s�[�N�ʐ�
(6) �X�s��-�X�s������
(1) NMR(�j���C����)�����@�Ƃ́H
�L�@���w�̌������t�������}�������A�V�����L�@�������̍\�������肷�邱�Ƃ́A���ɑ�ςȎd���ł����B�t���X�R�̒��ɉ��������Ă���̂����A�ǂ�����Č��߂�̂��Ƃ������Ƃ́A�ɂ߂Đ��I�Ȃ��Ƃ������̂ł��B�����̍��́A���̂悤�ȋ^��̉𖾕��@�́A���ׂāu���w������p�������͖@�v�ɗ����Ă��܂����B�Ⴆ�A�u�I�]������@�v��u��������@�v�Ȃǂ�p���āA���G�ȍ\���������q���A�\�����肪�e�ՂȒP���ȕ��q�ɕϊ����Ă����܂��B�����āA���������\��̒萫�����ɂ����A����̊��\��̗L���𐄑�����Ƃ����ߒ����J��Ԃ����Ƃɂ��A����ɖ����̂Ȃ��\�������肳��A����ɂ�������̔����Ńe�X�g����Ƃ��������Ƃ����x���J��Ԃ��A�悤�₭�������̍\�������肳��܂����B���̎�@�ɂ́A�c��Ȓm�������邱�ƂȂ���A��z���������Z�ʂ��K�v�Ƃ���܂����B�܂��A���̎�@�́A�\�������肷��̂ɐ��T�Ԃ��琔�J���A�Ƃ��ɂ͐��N�������邱�Ƃ�����܂����B
�������A�����ł́A�萫�����̌��ʂƉ��w�\���Ƃ̊֘A�t���́A�u�����I����v�Ɏ���đ����Ă��܂��B���̈Ӗ��ŁA���ׂĂ̌���̉��w�҂́A�u���͉��w�ҁv�ł���Ƃ����Ă��ߌ��ł͂���܂���B�������A����͌����āu���͉��w�ҁv�̐�含������ꂽ�Ƃ������Ƃł͂���܂���B�ނ���A���ׂẲ��w�҂��܂݂��ނ܂łɊg�債���Ƃ������Ƃł��B�u���O��������@(UV/VIS)�v���ŏ��ɏo�����A���q�ʂ𑪒�ł���u���ʕ��͖@(mass spectrometry�FMS)�v�A�����Č������Ă��錴�q�̐U�������m�ł���u�ԊO�����@(infrared spectroscopy�FIR)�v������ɑ����܂����B�����āA1950�N��̏I���ɂ́A�u�j���C����(nuclear magnetic resonance�FNMR)�����@�v������A�L�@���q�̍\������̐^�̊v�����n�܂�܂����B

�}.1 �v���g�������g��400 MHz�̒��`������(��㋳���w�ŎB�e)
�u�����I�ȕ��͖@�v�ɂ́A�u���w������p�������͖@�v�ɂ͂Ȃ������̒���������܂��B�܂��A���莎���̗ʂ��������ʂōς݁A�K�v�Ȃ�A���̎����𑪒�I����ɉ���ł��邱�Ƃ��������܂��B�܂��A����͐v���ɍs���āA�����ԂŏI��������̂������ł��B����ɁA�u���w������p�������͖@�v�����A���q�\���Ɋւ��鐔�����̏�A�X�y�N�g�����瓾���܂��B�����ł́A����̗L�@���w���x����uNMR�����@�v���A���q�\���̌�����@�Ƃ��āA�ǂ̂悤�ɗ��p����Ă��邩��������܂��傤�B
(2) NMR�̗��_
���ׂĂ̌��q�j�́A�d�ׂ������Ă��܂��B�������A���錴�q�j�ł́A���̓d�ׂ����q�j�̎���ŃX�s���^�����A���̊j�d�ׂ���邱�Ƃɂ���āA�������́u���C�o�Ɏq�v���܂��B�܂�A���q�j�͗z�d�ׂ������Ă���̂ŁA���ꂪ��]����Ǝ�������o�����ƂɂȂ�A���̉�]���錴�q�j�́A���������u�����Ȏ��v�̂悤�ȐU�镑��������̂ł��B���̂悤�ȋ������������q�j�̂����ŁA�L�@���w�ɍł��d�v�Ȃ��̂��A�u1H�v���u13C�v�̌��q�j�ł��B���q�j���X�s���������ǂ����́A�z�q�ƒ����q�̐��Ɉˑ����܂��B�L�@���w�̑�\�I�ȍ\�����q�ł���u12C�v��u16O�v�̌��q�j�́A�X�s���������Ȃ��̂ŁANMR�X�y�N�g���������܂���B���f���q�ł́A�V�R�ɑ��݂��邤����99%�ȏオ�j�X�s����L�����u1H�v�ł���ANMR�ł̊ϑ��ɍł��K�������f�ł��B
NMR�̌��ۂ́A���̐}.2�̂悤�ɂ��ċN����܂��B�X�s�������������q�j�́A���������̂ŁA���̂悤�Ȍ��q�j�́A1�́u�_���v�Ƃ��Č��Ȃ��܂��B�ʏ�A���́u�_���v�́A�ł���߂ɂ���������Ɍ����Ă��܂��B�������A�����Ɂu���͂ȊO������v�������ƁA���q�j�͊O������̕����ɉ����������A���邢�͋t�炤�����ɔz�܂��B�O������̕����ɉ������ѕ�(�����)�́A�t�炤���ѕ�(�����)�����A�G�l���M�[�I�ɂ킸���ɗL���ł���A���t��Ԃł́A���̃G�l���M�[�̒Ⴂ�L���Ȕz��(�����)�ɁA��葽���̌��q�j�����z���܂��B���̂��Ƃ��A�u�[�[�}������(Zeeman splitting)�v�Ƃ����܂��B
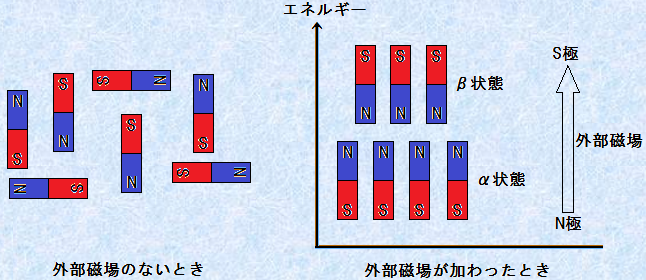
�}.2 �o���o���ɕ���ł���u�_���v�ɊO�����ꂪ�����ƁA�u�[�[�}������v���N����
�����āA�����Ɂu���W�I�g�̈�̓d���g�v���Ǝ˂���ƁA���q�j����N����āA�j�X�s������G�l���M�[���(�����)���獂�G�l���M�[���(�����)�ւƉ����グ���܂��B���̂Ƃ��̓d���g�̖����́A�j�X�s���̔z����ω�������̂ɕK�v�ȃG�l���M�[���������邱�Ƃł��B�܂��A���̂Ƃ����u���q�j���d���g�Ƌ����Ă���v�Ƃ����̂ŁA�u�j���C����(nuclear magnetic resonance)�v�̖��̂́A�������炫�Ă��܂��B�����āA�z�����ꂽ�d���g�̃G�l���M�[���A�M�Ƃ��Ď���ɓ�����A�����ꂽ���t���Ăь��ɖ߂�܂��B
�}.3 �����g���̓d���g���Ǝ˂���ƁA����Ԃ�������Ԃւ̌��q�j�̗�N���N����
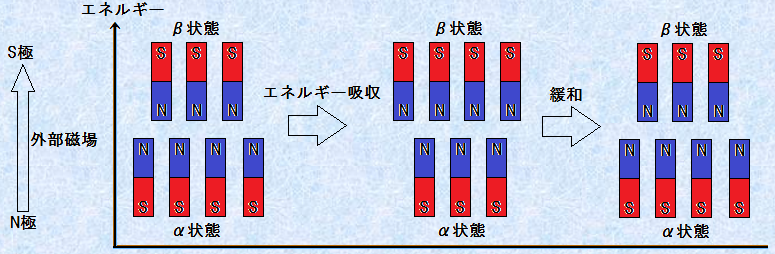
�O������ɉ����z��(�����)�ƁA�t�炤�z��(�����)��2�̏�Ԃ̃G�l���M�[���́A�������O������̋��x��A���q�̌�����Ԃɂ���Ă킸���ɈقȂ�܂��B�܂��A��ʓI�ɊO�����ꂪ�����قǁA���̃G�l���M�[�����傫���Ȃ�ANMR����̌��o���x�ƕ���\���ǂ��Ȃ�܂��B��ʓI�Ɏg�p�����͈͂̊O������̋���(1.4�`14 T)�ɑ��āA1H�̊j�X�s����2�̏�ԊԂ̃G�l���M�[���́A60�`600 MHz�̓d���g�����G�l���M�[�ɑ������܂��B����2�̊j�X�s���̃G�l���M�[�����A���w�҂ɐe���݂₷���ʂ̒P�ʂŕ\���ƁA25�`250 ��kJ/mol�Ƃ������ɏ����Ȓl�ɂȂ�܂��BNMR�����@�ł́A���̂킸���ȃG�l���M�[���ƕ��t��Ԃɖ߂�܂ł̎��ԕω����R���s���[�^�[�ŕ��͂��A�u�t�[���G�ϊ�(Fourier transform�FFT)�v�Ƃ�����@�ɂ��A�V�O�i�����x��\���X�y�N�g���ւƕϊ�����̂ł��B�L�@���w�҂́A����NMR�X�y�N�g����ǂނ��ƂŁA�L�@�������̍\�������肵�Ă����܂��B
|
�@ �X�s�������������q�j�����ꒆ�ɒu���ƁA�[�[�}�����N����B �A ���̃G�l���M�[���ɑ�������d���g���Ǝ˂���ƁA����Ԃ̌��q�j������Ԃ֗�N����B �B ���̃G�l���M�[���ƕ��t��Ԃɖ߂�܂ł̎��Ԃ𑪒肷�邱�ƂŁA�L�@�������̍\����������B |
(3) 1H NMR�̎�������
�@1H NMR�X�y�N�g���̑���́A��ʓI�Ɏ��̂悤�ɂ��čs���܂��B�܂��A���肵�������~���O�����̎������������A��0.4 mL�̗n�}�ɗn�����āA�O�a5 mm�̃K���X��(NMR�`���[�u)�ɓ����܂��B�n�t�ł́A�������q�͑��ʂ̗n�}���q�̒��ɕ��U���A���ꂼ�ꃉ���_���ɓ������Ȃ��瑶�݂��܂��BNMR�́A����������Ԃ̏W���̕��ω����ꂽ���̂��ϑ����Ă��܂��B400 MHz�̑��u�ő��肷��Ƃ��ɂ́A1 mg���x�̎����������ł��A�\����1H NMR�X�y�N�g���������܂��B�����ł́A�������������A�n�}�ɑ��Ċ��S�ɍ��a���Ă��邱�Ƃ��d�v�ł��B

�}.4 ������������n�}�ɗn�����āANMR�`���[�u�ɓ����
�������A�n�}���܂�1H���܂ނ��߁A���ʂ̗L�@�n�}�ɗn�����đ��肷��ƁA�n�}�̃X�y�N�g�����傫���ϑ�����Ă��܂��A���肵�����������̃X�y�N�g��������ɖ�����Ă��܂��܂��B1H NMR�ŗ��z�I�ȗn�}�́A�v���g�������̍\���Ɋ܂܂��A����ɍ����łȂ��A���_���Ⴍ�A���ɐ��ł���A�����ĕs�����ł��邱�Ƃł��B�l�����Y�fCCl4�́A����Ɏ������\���ɗn����Ȃ�A���z�I�ȗn�}�ł��B�܂��A�d���f���N�����z����CDCl3�́A�ł��L���p������n�}�ł��B���̒��ɕs�����Ƃ��Ċ܂܂�邱�Ƃ������N�����z����CHCl3���琶����X�y�N�g�����A�r�������W�Q�����邱�Ƃ͂قƂ�ǂ���܂���B�قƂ�ǂ��ׂĂ̗n�}���A98�`98.8%�̓��ʑ̏��x(D���q%)�ŏd���f�����ꂽ�`�œ���ł��܂��B�܂��A�����1H NMR�X�y�N�g���̑���ŗp������n�}�́A��ʓI�ɕ��_���Ⴂ�̂ŁA���肪�I�������n�}�𗯋����āA������������������邱�Ƃ��ł��܂��B
(4) ���w�V�t�g
�@���f���q���܂�1�̉������ɑ��āA������1�{�̋z�����ϑ�������ł͂���܂���B���q���̂��ׂĂ̈قȂ鐅�f�́A���ꂼ��قȂ�ʒu�ɋz���������A�e���f�ɑ��鋤���g��(�d���g�̃G�l���M�[�ƑΉ�����)�́A���̐��f�ɒu���ꂽ�u���w�I���v�ɋ����ˑ�����̂ł��B�����ŁA���q���Ő��f�̒u���ꂽ���w�I���̈Ⴂ�ɂ�鋤���g���̕ω����A�u���w�V�t�g(chemical shift)�v���Ăт܂��B���w�I���̈Ⴂ���A��̂ǂ̂悤�ɉ��w�V�t�g�ɉe������̂��A�l���Ă݂܂��傤�B
���q���̐��f�́A���q�O���ɂ���āA���߂�ꂽ������̋�Ԃ��߂Ă��܂��B�����ɊO������B0����������ƁA�d�q�͂��̋�Ԃ̒��ʼn~�^�����N�����A�O������ɋt�炤�悤�Ȏ���Bi��U�N���܂��B
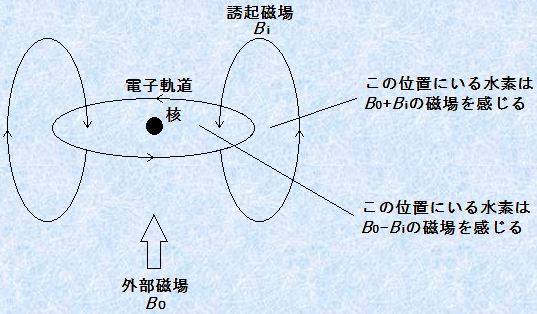
�}.5 �~�^��������d�q�́A�j�́u�������Օ����ʁv�������炷
���������āA���f�������鐳���̎���(B0�}Bi)�́A���q���̂��ׂĂ̈قȂ鐅�f�ɂ��ď������قȂ�A�����g�����܂��Ⴄ�͂��ł��B�U�N���ꂽ����̂��߂ɁA�قƂ�ǂ̐��f���A�O����������ア����(B0-Bi)�������܂��B���̂悤�ȂƂ��A���̐��f�́u�Օ�(shielding)�v����Ă���Ƃ����܂��B�Օ��̒��x�́A�~�^��������d�q�́u���x�v�Ɉˑ����A��ʓI�ɓd�q���x���傫���قǁA�Օ����ʂ͋����Ȃ�܂��B���ׂĂ̗L�@�����̎����u���������ہv�́A���̌��ʂɂ���Đ��������̂ł��B
�܂��A1H NMR�ł́A���ʒu��������̃V�O�i���ɑ��鑊�Έʒu�Ŏ����K�v������A�����g���̍���1/106 (ppm)��P�ʂƂ��ĕ\������܂��B������Ƃ��ẮA��ʓI�Ɂu�e�g�����`���V����(Si(CH3)4�FTMS)�v���I��܂��B���̉������ɂ́A�������̒����A���Ȃ킿�A���w�I�s�����A���C�������A������(b.p.27��)�A����ё啔���̗L�@�n�}�ւ̉n���Ƃ����D�ꂽ����������܂��BTMS��12�̐��f���q�́A���w�I�ɓ����ł���A���w�V�t�g�ɂ�����1�{���Ō����̂ŁA���̉s���V�O�i������_�Ƃ��ėp����ƁA���ɓs�����ǂ��̂ł��B������ATMS�͒ᕦ�_�ł��邩��A����I����͗e�Ղɏ����ł��܂��B�Ȃ��A�d���f���N�����z����CDCl3�ɗ\��TMS��0.05wt%���x�ܗL���ꂽ���̂��s�̂���Ă���̂ŁATMS�𑪒�̓x�ɗp�ӂ���K�v�͂���܂���B
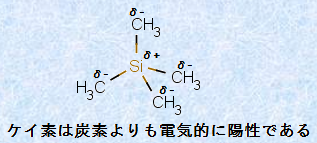
�}.6 �u����������v�Ƃ��ėp������TMS
���w�V�t�g�ɂ����āATMS��1�̉s���z���s�[�N�������܂����A����́A�قƂ�ǂ��ׂĂ̗L�@�������̃v���g�������u������v�ŋz�����܂��B���Ȃ킿�ATMS�̐��f�͓d�q���x�����ɍ����A�����Օ�����Ă���̂ŁA���ʂ̗L�@�������ɔ�ׂ�ƁA�u��������v���K�v�ƂȂ�̂ł��B���������āA���̐}.7�̂悤�ɁATMS�͈�ʓI�ɃX�y�N�g���̍ł��E��(0 ppm)�Ɍ���܂��B
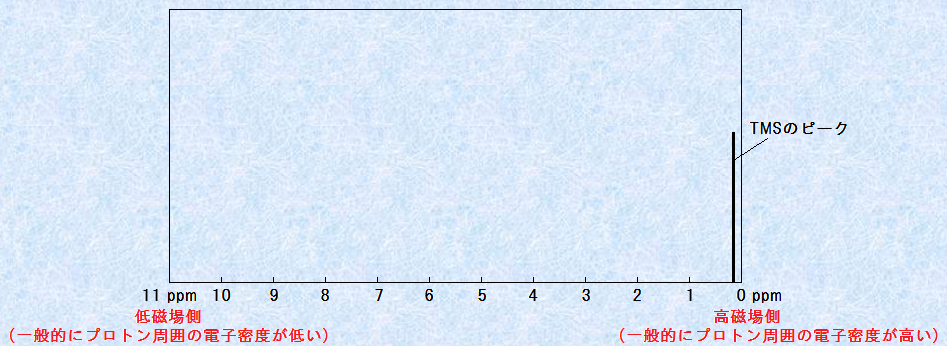
�}.7 1H NMR�ړx
�}.7�̂悤��1H NMR�ړx�����A�E�[��0 ppm��TMS�s�[�N���߂܂��傤�B�����̗L�@�������́A��ʓI�Ƀv���g�����͂̓d�q���x��TMS�����Ⴂ�̂ŁA�u�Ꭵ�ꑤ�v�ɋ��s�[�N�������܂��B���������āA���̈ʒu���v���X�́u���l�v�Ŏ����ɂȂ��Ă��܂��B�Ⴆ�A����1.00�Ƃ́ATMS�s�[�N����1 ppm�����A�Ꭵ��̈ʒu�ɋ��s�[�N������邱�Ƃ��Ӗ����Ă��܂��B�܂�A400 MHz�̓d���g���Ǝ˂��Ȃ��瑪�肵���ꍇ��1 ppm�Ƃ́ATMS����400 Hz�����Ꭵ��̈ʒu���w���܂��B
�܂�A�v���g���̉��w�V�t�g�Ƃ́ATMS����Ƃ����v���g��������l�̂��ƂȂ̂ł��B���w�V�t�g�ƌĂ�闝�R�́A���l���A���̐��f���q����芪�����w�I���̈Ⴂ�ɂ��ω�����ʂ�����ł��B�܂��A���̉��w�V�t�g�́A�u�O������̋����v�ɐ���Ⴗ��l�Ȃ̂ŁA�u�����g���v�ɂ�����Ⴕ�A�u���葕�u�̈Ⴂ�v�ɖ��W�̗ʂł��B���������āA�قȂ镪�q�ł����Ă��A�������w�I���ɂ��鐅�f�́A�ǂ�ȑ��葕�u�Ōv�����Ă��A�������w�V�t�g�������܂��B��d�����ɕt�������`����(-CH3)�̋z���́A�ǂ���قƂ�Ǔ����ʒu�ł��邵�A�F�������f�́A����ɓ����I�Ȉʒu�ŋz�����N�����܂��B���̕\.1�ɁA�l�X�Ȑ��f�̋z���ʒu���܂Ƃ߂Ă݂܂����B�������A�\�Ɏ����ꂽ�l�́A�T���̒l�ł���A�����Ŏ��������l�́A�O���܂Ŗڈ��ł���Ƃ������Ƃɒ��ӂ��Ă��������B
�\.1 1H NMR�̗l�X�ȉ��w�V�t�g
|
|
1H�̎�� |
�� (ppm�l) |
|
���`���v���g�� -CH3 |
CH3-CR3 |
����0.8�`1.3 |
|
CH3-CR2X |
����0.8�`2.0 |
|
|
CH3-C=C |
����1.4�`2.4 |
|
|
CH3-C=O |
����1.8�`2.8 |
|
|
CH3-�n���Q�� |
����2.2�`4.3 |
|
|
CH3-�F���� |
����2.2�`3.0 |
|
|
CH3-NR2 |
����2.2�`3.9 |
|
|
CH3-OR |
����3.3�`4.3 |
|
|
���`�����v���g�� -CH2-
|
R-CH2-CR3 |
����1.2�`1.7 |
|
R-CH2-CR2X |
����1.2�`2.0 |
|
|
R-CH2-C=C |
����1.8�`2.4 |
|
|
R-CH2-C=O |
����2.0�`2.8 |
|
|
R-CH2-�n���Q�� |
����2.2�`4.4 |
|
|
R-CH2-�F���� |
����2.4�`3.3 |
|
|
R-CH2-NR2 |
����2.4�`3.8 |
|
|
R-CH2-OR |
����3.4�`4.4 |
|
|
���`���v���g�� -CH- |
R2-CH-CR3 |
����1.4�`1.6 |
|
R2-CH-CR2X |
����1.5�`2.0 |
|
|
R2-CH-C=O |
����2.3�`3.0 |
|
|
R2-CH-�n���Q�� |
����4.0�`5.8 |
|
|
R2-CH-�F���� |
����2.7�`3.3 |
|
|
R2-CH-NR2 |
����2.6�`4.2 |
|
|
R2-CH-OR |
����3.5�`5.3 |
|
|
���d�����Ɍ������� �v���g�� |
C��C-H(���[�A���L��) |
����1.8�`3.0 |
|
C=C-H(���[�A���P��) |
����4.6�`6.4 |
|
|
C=C-H(�����A���P��) |
����5.7�`7.7 |
|
|
�F�����v���g�� |
����6.5�`8.5 |
|
|
�w�e���F�����v���g�� |
����5.7�`9.3 |
|
|
O=C-H(�A���f�q�h) |
����9.0�`10.0 |
|
|
-COOH(�J���{���_) |
����10.0�`13.0 |
���w�V�t�g�Ɋւ��āA�u�d�C�A���x�v�̊T�O�́A������x�܂ł͐M���ł��铱���ƂȂ�܂��B�Ⴆ�ATMS�̃s�[�N���A�قƂ�ǂ̗L�@����������������̈ʒu�Ɍ����̂́A�P�C�fSi�͒Y�fC�ɑ��ēd�C�I�ɗz���ł���ATMS�̃v���g������̓d�q���x�������Ȃ邩��ł��B����̂ɁA�����̃v���g���͍��x�ɎՕ�����Ă���A�����̃s�[�N�͍����ꑤ�Ɍ����̂ł��B�u�d�C�A���x�v�Ɓu�v���g�����͂̓d�q���x�v�̊T�O���g�p����A���w�V�t�g�ɂ��āA�����̗L���Ȑ�����������Ƃ��ł��܂��B�܂�A�d�q�������̊�́A�אڂ��鐅�f�̓d�q���x��ቺ������̂ŁA���f���q�j���Օ�������ʂ��������܂��B���ʂƂ��āA���̐��f�̉��w�V�t�g�́A��r�I�Ꭵ�ꑤ�Ɍ���邱�ƂɂȂ�̂ł��B��Ƃ��āA���̕\.2�ɁA�n���Q���Œu�����ꂽ�A���J���̉��w�V�t�g�������܂��B���̕\����́A�u����̓d�C�A���x���傫���قǁA���f���q�j�ߖT�̓d�q�����������t���āA���ʒu���Ꭵ�ꑤ�Ɉڂ邱�Ƃ�������܂��B�܂��A�u�d�q���x���Ⴂ�v�Ƃ������Ƃ́A�u�_���x�������v�Ƃ������Ƃɂ��Ȃ�̂ŁA���������������A�_���x�̍������f�قǁA���̉��w�V�t�g�́A�Ꭵ�ꑤ�Ɍ����Ƃ������Ƃɂ��Ȃ�܂��B
�\.2 �n���Q���Œu�����ꂽ�A���J���̉��w�V�t�g
|
|
X��Cl |
X��Br |
X��I |
|
CH3X |
3.1 |
2.7 |
2.2 |
|
CH2X2 |
5.3 |
5.0 |
3.9 |
|
CHX3 |
7.3 |
6.9 |
5.4 |
�@����ɁA�\.1�̉��w�V�t�g�ɂ��ƁA�u���d�����v��u�F���v���\������Y�f���q�Ɍ����������f���q�́A�u�O�a�Y�f�v�Ɍ����������̂��A��ʓI�ɒᎥ�ꑤ�Ɍ����Ƃ������Ƃ�������܂��B�m���ɁA�A���P����sp2�Y�f��A���L����sp�Y�f�́A�A���J����sp3�Y�f��荂��s���������A�߂��̐��f����d�q���������A���̐��f�̎Օ�����߂���ʂ������܂��B�������A���̌��ʂ��l�����Ă��A�F���̃v���g���Ȃǂ́A�\�z�ȏ�ɒᎥ�ꑤ�̉��w�V�t�g���܂��B
���̌����́A�~�^���������d�q�ɂ���Đ������U�N����ɂ���܂��B�Ⴆ�A�F���Ɍ����������f�̉��w�V�t�g�́A�\�z�ȏ�ɒᎥ�ꑤ�Ɍ���܂����A����́A�O������B0���A�F��������d�q�̉~�^����U�N���A���ꂪ�O������B0�Ƌt�����̗U�N����Bi�������邩��ł��B���f���������Ă���F���̊O���ł́A�U�N����Bi�́A�O������B0�����߂�悤�ɍ�p���܂��B����̂ɁA�F���v���g���́A�Ⴂ����ł������N����̂ł��B�u�F���v���g���v�̉��w�V�t�g��=6.5�`8.5�́A���ɏd�v�Ȓl�ł���A���͈̔͂ɃV�O�i��������A��ʓI�ɖF���Ɍ����������f�����݂���Ɣ��f���邱�Ƃ��ł��܂��B
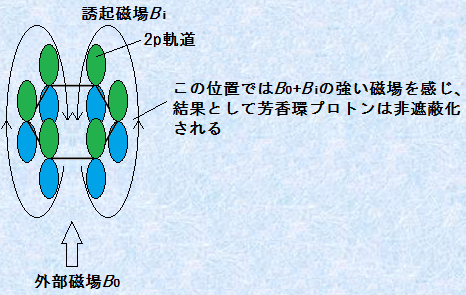
�}.8 �u�F���v���g���v�̔�Օ�������
(5) �s�[�N�ʐ�
�X�y�N�g�����̃V�O�i���̑��ΓI�ȋ��x�𑪒肷�邱�Ƃɂ��A����̃V�O�i����^���鐅�f�̐��𐄒肷�邱�Ƃ��ł��܂��B���Ȃ킿�A�e�V�O�i���́u�s�[�N�ʐ�(peak area)�v�́A���̃s�[�N�ɑ����Ă���u�v���g���̐��v�ɐ���Ⴗ��̂ł��B�s�̂�NMR���u�ɂ́A���ׂēd�q���ώZ�v���t�����Ă���A���̌v�Z�ɂ���āA�s�[�N��Ɂu�ϕ��Ȑ�(integration line)�v���������܂��B���̋Ȑ��̐��������̍����̔䗦���A���傤�ǃs�[�N�ʐς̔䗦�ɂȂ�܂��B��Ƃ��āA���̐}.9�ɁA4,4-�W���`��-2-�y���^�m����1H NMR�X�y�N�g���������܂��B
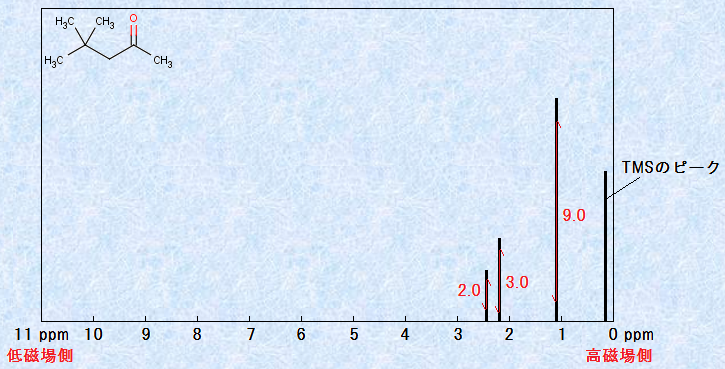
�}.9 4,4-�W���`��-2-�y���^�m����1H NMR�X�y�N�g��
4,4-�W���`��-2-�y���^�m���ɂ́A���v14�̐��f������܂����A���̐��f�́A�d�q�I�����猩��ƁA��������3��ނ�������܂���B���Ȃ킿�A���`����(-CH3)��3�̐��f�ƃ��`������(-CH2-)��2�̐��f�́A���ꂼ�ꉻ�w�I�ɂ������w�I�ɂ������ł���Atert -�u�`����(-C(CH3)3)��9�̐��f���܂��A���w�I�ɂ������w�I�ɂ������ł���Ƃ������Ƃł��B����āA4,4-�W���`��-2-�y���^�m���ɂ́A3�{�̃V�O�i��������A���̐ϕ��̔䗦�́A9�F3�F2�ł��邱�Ƃ������Ă��܂��B���w�V�t�g�̈ʒu(���l)�ɉ����āA�e�V�O�i���̐ϕ��l(���f�̐�)��������A�P���ȉ������̃V�O�i���̋A���͗e�ՂɂȂ�܂��B�������A�قƂ�ǂ̏ꍇ�ŁA���ꂾ���ł̓V�O�i���̋A��������ł���ꍇ�������̂ł��B���ꂩ���������i��ŁA�u�X�s��-�X�s�������v���l�����܂��傤�B
(6) �X�s��-�X�s������
NMR�����@�̗L�p���́A�P���ɗL�@���������̐��f�����o�ł���Ƃ��������ł͂���܂���B����܂ŏq�ׂĂ����悤�ɁANMR�����@�ɂ��A���q���̈قȂ��ނ̐��f�̐������肷�邱�Ƃ��ł��A�����̐��f����߂鉻�w�I���̊T����m�邱�Ƃ��ł��܂��B�������Ȃ���A����ɑ����̏���NMR���瓾����@������̂ł��B
�قƂ�ǂ�1H NMR�X�y�N�g���́A�����ɂ́A�}.9�̂悤�ɒP���ł͂���܂���B��ʓI��1H NMR�X�y�N�g���ł́A���ׂĂ̐��f��1�{���́u�V�O�i��(s�Fsinglet)�v�ł���Ƃ͌��炸�A���G�ȃs�[�N�ƂȂ��Č���邱�Ƃ������̂ł��B���E���G�`��CH3CH2I�́A���̍D��ł��B�܂��\�z�ł��邱�Ƃ́A�X�y�N�g���́A���`����(-CH3)��3�̓����Ȑ��f�ƃ��`������(-CH2-)��2�̓����Ȑ��f�ɂ��A������2�{�̃V�O�i������Ȃ邾�낤�Ƃ������Ƃł��B����ɁA�\.1���Q�Ƃ���A�e�s�[�N�����������ǂ̈ʒu�Ɍ���邩�����ł��܂��B�������A���ۂ̃X�y�N�g���́A�\�z�Ƃ͂��Ȃ�قȂ�܂��B�\�z�����ʒu�ɃV�O�i���͂�����̂́A�����͂������̐�����ł��Ă��āA�}.9�̂悤�ɁA���ׂẴV�O�i�����u�V���O���b�g�v�ł���̂Ƃ͈Ⴂ�܂��B
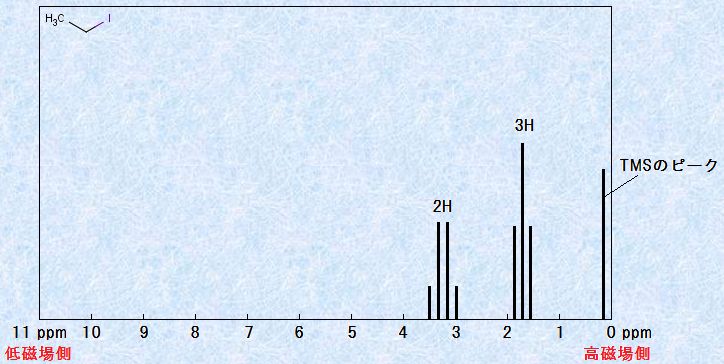
�}.10 ���E���G�`��CH3CH2I��1H NMR�X�y�N�g��
���`�����f(-CH3)�̃V�O�i���́A����1.85�ɒ��S������1�F2�F1�̋��x���3�{���Ō���A���`�������f(-CH2-)�́A����3.2�𒆐S�Ƃ���1�F3�F3�F1�̋��x���4�{���Ō���Ă��܂��B�e���̊Ԋu�͂��ׂē������A���̒l��7.6 Hz�ł��B���{���̐��ɕ����V�O�i���́A�u�_�u���b�g(d�Fdoublet)�v�E�u�g���v���b�g(t�Ftriplet)�v�E�u�J���e�b�g(q�Fquartet)�v�Ȃǂƕ\����܂��B�Ȃ����̂悤�ȕ�������̂ł��傤���H�܂��A���������w�҂́A����̏����ǂ̂悤�ɗ��p�ł���̂ł��傤���H
�\.3 �s�[�N�̌`�ƕ����{��
�܂��A���E���G�`��CH3CH2I�̃��`�����f(-CH3)�́u�g���v���b�g�v�ׂĂ݂܂��傤�B���̂��߂ɂ́A�܂��ׂ̃��`�������f(-CH2-)�ɒ��ڂ��܂��B���`������(-CH2-)��2�̓����Ȑ��f�́A���ꂼ��u���v�܂��́u���v�̃X�s���������Ă��܂��B�����ŁA�����X�s���̈قȂ�g�ݍ��킹���A4�g�ł��܂��B���Ȃ킿�A(��,��)�E(��,��)�E(��,��)�E(��,��)��4�ʂ�ł��B���`�����f(-CH3)�������鐳���̎���́A���̑g�ݍ��킹���Ƃɑ����ω����܂��B���������āA���`�����f(-CH3)�́A�ׂ̃��`�������f(-CH2-)�̃X�s���̑g�ݍ��킹�ɉ����ĕω������O��������A�����̎���Ƃ��Ċ�����̂ł��B��������ƁA4�g�̃X�s���̑g�ݍ��킹�ɑΉ����āA4�{�������������Ɏv���܂��B�������A(��,��)��(��,��)�̑g�ݍ��킹�������ł���A�����̐����̎���ނ̂ŁA���ʂƂ��āA1�F2�F1�̔䗦��3�{��(��,��)�F(��,��)�{(��,��)�F(��,��)����Ȃ郁�`�����f�V�O�i���������̂ł��B���̂Ƃ��A���`�����f(-CH3)�́A�ׂ̃��`�������f(-CH2-)�Ɓu�X�s��-�X�s������(spin-spin coupling)�v���Ă���Ƃ����܂��B���̊Ԋu�́A�u�����萔(coupling constant�F�L��J�Ŏ���)�v�ƌĂ�A�uHz�v�̒P�ʂő����܂��B���E���G�`��CH3CH2I�̏ꍇ�A���̊Ԋu��J��7.6 Hz�ł��B
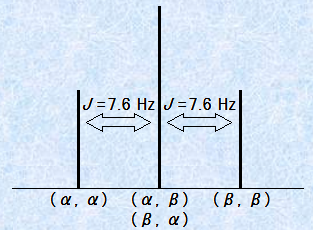
�}.11 ���E���G�`��CH3CH2I�̃��`�����f(-CH3)�̃V�O�i��
���x�́A���E���G�`��CH3CH2I�̃��`�������f(-CH2-)�́u�J���e�b�g�v�����߂��Ă݂܂��傤�B�ׂ̃��`����(-CH3)�ɂ́A3�̓����Ȑ��f�̊j�X�s���ɂ��āA(��,��,��)�E(��,��,��)�E(��,��,��)�E(��,��,��)�E(��,��,��)�E(��,��,��)�E(��,��,��)�E(��,��,��)��8�ʂ�̑g�ݍ��킹������܂��B�������Ȃ���A���̂���3�̃X�s���̑g�ݍ��킹�������Ȃ��̂��A2�g(��,��,��)�E(��,��,��)�E(��,��,��)�����(��,��,��)�E(��,��,��)�E(��,��,��)����܂��B���������āA���`�������f(-CH2-)�́A�ׂ̃��`�����f(-CH3)�̈قȂ�4�ʂ�̑g�ݍ��킹�ɂ���ĕω���������ɒu����邱�ƂɂȂ�A���̃X�y�N�g���́A1�F3�F3�F1�̔䗦�̎l�{��(��,��,��)�F(��,��,��)�{(��,��,��)�{(��,��,��)�F(��,��,��)�{(��,��,��)�{(��,��,��)�F(��,��,��)�Ƃ���̂ł��B���`�������f(-CH2-)�́A���`�����f(-CH3)�����`�������f(-CH2-)�ɃX�s���������Ă���̂Ɠ���̌����萔J��7.6 Hz�������A���`�����f(-CH3)�ƃX�s���������Ă��܂��B
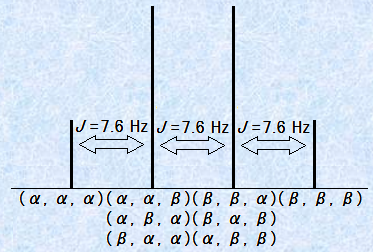
�}.12 ���E���G�`��CH3CH2I�̃��`�������f(-CH2-)�̃V�O�i��
�@1�̐��f�̃V�O�i���̕���́A�אڂ��Ă��鐅�f�ɂ���Ĉ����N������A�����V�O�i���̐��́A�����̐��f�̐��ɂ���Č��肳��܂��B�܂�A�אڂ���1�̐��f�́A�u�_�u���b�g�v�������A�אڂ���2�̓������X�s���������Ă��鐅�f�́A�u�g���v���b�g�v��������̂ł��B�אڂ���Y�f�Ɂun�v�̓����Ȑ��f������ꍇ�A�����̐��f�́A�un�{1�{�v�̃V�O�i���������A���̐��̊Ԋu��J Hz�ƂȂ�܂��B����́A��ʓI�Ɂun�{1�̋K���v�Ƃ��Ēm���Ă��܂��B
�e���̋��x��́A�X�s�������̑��萅�f�̉\�ȃX�s���̑g�ݍ��킹����͂��邱�Ƃɂ�茈��ł��܂��B���邢�́A���̐}.13�̂悤���u�p�X�J���̎O�p�`(Pascal's triangle)�v���g���Ă����o�ł��܂��B���̎菇�ł́A�e�����͂������2�̐����̘a�ɂȂ��Ă���A�����̐������A�e���̋��x���\���悤�ɂȂ�܂��B�p�X�J���̎O�p�`���g���ANMR�V�O�i���̐��̋��x�䂪�����ɕ�����̂ł��B�������A�����ŋ��߂���̂́A����1�̃V�O�i���̒��ł̕������̑��ΓI���x�ł����āA���q���̕ʂ̐��f�̃V�O�i�����x�ł͂Ȃ����Ƃɒ��ӂ��K�v�ł��B

�}.13 �u�p�X�J���̎O�p�`�v���g���A�e���̋��x�䂪���߂���
�E�Q�l����
1) H.�n�[�g/L.E.�N���[��/D.J.�n�[�g �����u�n�[�g��b�L�@���w�v�|����(1986�N���s)
2) ���[�g�����h�E�W���[���Y�u�W���[���Y�L�@���w(��)�v�������w���l(2000�N���s)
3) �V���o�[�V���^�C��/�E�F�u�X�^�[/�L�[���� �����u�L�@�������̃X�y�N�g���ɂ�铯��@-MS, IR, NMR, UV�̕��p-�v�������w���l(2006�N���s)
4) �R�c���v�u�j���C���@�Ǝ��ʕ��͖@�̊�b�F�L�@�������̎�ށE�\�����ǂ̂悤�ɒ��ׂ邩�H�v���w�Ƌ���70��4��(2022�N)